
1. INTRODUCTION
Agricultural crop production is mainly dominated by three crops worldwide (maize, wheat, and rice) which account for about 90% of cereals [1]. Among these, wheat is one of the most important cereal staple food crops and it is grown in more than 25% of cultivated land areas worldwide. Plant growth-promoting bacteria (PGPB) are intricately associated with roots in the rhizosphere. PGPB enhances plant growth through phosphate solubilization, siderophore production, nitrogen fixation, phytohormone production such as indole-3-acetic acid, cytokinin, and gibberellic acid and through acidification [2,3]. These PGPB, in association with mycorrhizae, could be employed to enhance the nutrient content, grain yield in wheat, and soil health. PGPB and other neighboring rhizosphere microorganisms alter the physiological, metabolic, and morphological properties of plants to increase overall growth, in addition to N-assimilation and N-availability [4]. Rhizospheric plant-associated microbiota can be considered as an extended plant genome that plays a significant role in regulating cellular homeostasis [5]. Similarly, PGPB inoculation leads to metabolic alterations that favor mineral nutrient availability and uptake, respiration, and proliferation of roots [6,7]. Understanding the molecular mechanisms underlying PGPB and arbuscular mycorrhizal fungi (AMF)-mediated plant growth promotion requires a comprehensive analysis of proteomic and metabolomic changes in plant tissues. Grain tissue proteomics analysis in wheat provides insights into seed development, storage protein accumulation, and stress response mechanisms that directly influence grain quality and yield [8]. Root tissue metabolomics reveal changes in primary and secondary metabolite profiles that reflect altered nutrient uptake, stress tolerance, and plant-microbe communication [9]. Several studies have demonstrated that microbial inoculation significantly modulates wheat grain proteome, thus affecting proteins involved in carbohydrate metabolism, defense responses, and cellular homeostasis [10,11]. Similarly, metabolomic analyses have revealed that PGPB- and AMF- treatments change the accumulation of key metabolites such as amino acids, sugars, and various secondary metabolites, which play crucial roles in plant growth regulation and stress adaptation [12,13]. Differential abundance of metabolites in plants subjected to microbial treatment can be assigned to known regulatory metabolic pathways and was found to be correlated with the phenotype/trait changes [13,14]. A few studies have investigated the effect of PGPB and mycorrhiza on metabolomic changes in plants. These studies inferred that the differentially regulated metabolites have a role in plant growth regulation, biotransformation, as well as stimulation [10-12,15]. In addition to metabolomics, proteomic techniques provide a unique angle in deciphering the mechanistic details of plant–microbe interactions [16].
In our previous studies, we have reported that dual inoculation of wheat plants with PGPB and AMF substantially improves plant growth, grain yield, and micronutrient biofortification under greenhouse and field conditions [17-19]. In our 1st-year field trial, it was reported that plants treated with CP4 (native PGPB) and AMF not only enhanced grain yield and biofortification in wheat but also modulated various proteins and metabolites [18,20]. AHP3, a non-native PGPB (GenBank accession no. MN607032), also showed prominent plant growth-promoting (PGP) activity in the 1st-year field trial. Therefore, combined treatment of native CP4 and non-native AHP3 along with AMF was tested in the 2nd-year field trial [19]. Based on plant growth, yield, nutrition, and soil fertility parameters, CP4+AHP3+AMF proved to be an effective microbial consortium with excellent application prospects. However, the mechanistic aspects underlying the enhanced growth and grain yield due to CP4+AHP3+AMF inoculation were not studied previously. Here, we investigated the influence of microbial consortia treatment on the proteomic profile of wheat grain tissue and the changes in the metabolomic profile of grain and root. We hypothesized that (a) treatment with CP4+AHP3+AMF will differentially affect the regulatory proteins and metabolites involved in growth and grain yield, and (b) we will be able to identify metabolic pathways involved in improved nutrient acquisition.
2. MATERIALS AND METHODS
2.1. Background Experimental Design
The 2nd-year field trial, which was part of a 2-year field study, was conducted with HD-3086 (Pusa Gautami) wheat variety, which is widely cultivated by farmers in the North-Western zone of India. Two Bacillus spp. PGPB, spp. CP4 (GenBank accession no. MN607032) and AHP3 (GenBank accession no. MN607035), and Symbion-VAM Plus™, containing mainly Glomus spp. (https://tstanes.com/domestic-crop-care/bio-fertilizers/symbion-vam-plus), were used for inoculation. Inoculation was performed at the seed stage before sowing and during growth stages in a randomized split-block design field experiment. For non-inoculated wheat samples (control), equivalent distilled water was applied instead of microbial inoculum. All samples were analyzed with three biological replicates collected from respective experimental blocks. Plant growth and yield-related parameters, total chlorophyll, grain and root nitrogen, phosphorus, iron, zinc and total phenolic content, and oxidative markers in grain tissue were analyzed as reported in Yadav et al. [18].
To study the proteomic and metabolomic changes in wheat plants in response to various microbial inoculation under field conditions, four treatments were investigated co et al. [18].mpared to the control: (1) CP4-inoculated plants, (2) AMF-inoculated plants, (3) CP4+AMF-inoculated plants, and (4) CP4+AHP3+AMF-inoculated plants.
2.2. Proteomic Analysis of Wheat Grains
Plant samples (grain tissue) collected in biological duplicates at the harvesting (maturity) stage were immediately stored in liquid nitrogen. An equal amount of wheat grains from each sample was pulverized in liquid nitrogen. Wheat grain proteins were extracted as per Yang et al. [8]. Briefly, wheat grain tissue (100 mg) was suspended in an extraction buffer comprising 0.5 M Tris-HCl (pH 7.5), 0.7 M sucrose, 50 mM ethylenediaminetetraacetic acid (EDTA), 0.1 M KCl, and 1% (w/v) 1,4-bis(sulfanyl)butane-2,3-diol (DTT). Further, phenylmethylsulfonyl fluoride (PMSF), a serine protease inhibitor, was added (0.1 mM PMSF, pH 7.5), followed by incubation for 30 min at 4°C with intermittent shaking. The mixture was centrifuged for 30 min at 8,000 rpm, followed by rinsing of the pellet in acetone with 0.2% w/v DTT. The pellet was resuspended in guanidine HCl (6M, pH 8.0). The total protein was estimated with the BCA Protein Assay Kit (Pierce™). Global proteome analysis of the protein samples was carried out by VProteomics (https://www.vproteomics.com/) as reported in Yadav et al. [20]. Briefly, 25 μL of protein sample was reduced with Tris (2-carboxyethyl) phosphine hydrochloride TCEP-HCl (Tris(2-carboxyethyl)phosphine) followed by alkylation and digestion with 50 mM iodoacetamide and trypsin, respectively. A C18 silica column was used for desalting the sample followed by drying. The pellet was dissolved in acetonitrile and formic acid.
2.3. Peptide Analysis and Data Processing
Proteome analysis was performed on ThermoFisher-Q Exactive coupled with EASY-nLC 1000. A PicoFrit column (C18) was used to resolve the peptide mixture, and Proteome Discoverer (v2.2) was used to analyze the raw data. Protein digestion was done using trypsin/P. Protein abundance values were quantified after performing normalization against the total peptide amount in each replicate. The cut-off used was 0.01 for peptide spectrum match and protein false discovery rate (FDR). MS Amanda 2.0 with default parameters and T. aestivum as reference proteome was used, with two replicates having at least two peptide hits selected for protein quantification and identification. The proteins were annotated using the UniProt ID mapping function. Differentially abundant proteins were determined after normalizing the peak intensity, and log2 transformation was performed. Further, proteins having a significant change in abundance were represented by P < 0.05 and absolute Log2 fold change ≥1.
2.4. Wheat Metabolites Extraction and Sample Preparation for Gas Chromatography-Mass Spectrometry (GC-MS) Analysis
Wheat grain and root tissues were collected in biological triplicates at the harvesting stage, and metabolomic profiling was performed for each group (control and PGPB/AMF-treated). Immediately after plant harvesting, the samples were stored at −80°C, followed by lyophilization in liquid nitrogen for 72 h. Lyophilized grain tissue (100 mg) was used for GC-MS-based metabolomic profiling as per Yadav et al. [18] and Lisec et al. [21]. Briefly, lyophilized wheat grain tissue (100 mg) was suspended in pre-chilled methanol. Subsequently, adonitol was added as an internal standard. The mix was centrifuged for 20 min at 10,000 rpm. The supernatant was collected in an Eppendorf tube, and chloroform and sterile water were added. This step was repeated again. The supernatant was transferred to a 15 mL tube, followed by drying and derivatization, as per Yadav et al. [18]. Subsequently, the samples were mixed with an index standard mixture of N-Methyl-N-(trimethylsilyl) trifluoroacetamide. The filtrate was collected in a GC-MS vial and submitted to the central instrumentation laboratory (CUP), Bathinda, India. Metabolite peak identification and normalization were performed as described by Yadav et al. [18].
2.5. Metabolomics Data Analysis
GC-MS analysis was performed using a GC-MS-QP2010 Ultra system (Shimadzu Corp., Kyoto, Japan). The analytical conditions were optimized with column and injector temperatures maintained at 70°C and 250°C, respectively. Direct injection mode was employed throughout. The carrier gas flow parameters were set as follows: Pressure at 66.7 kPa, column flow rate of 1.07 mL/min, linear velocity of 37.9 cm/s, and purge flow rate of 3 mL/min. The mass spectrometer operating conditions included an ion source temperature of 200°C, an interface temperature of 260°C, and a solvent cut time of 5.5 min. Metabolite profiling was conducted across a temperature gradient of 50–330°C with helium as the carrier gas at 1 mL/min flow rate and an injection volume of 0.5 μL. MS data acquisition was performed at a scan rate of 2 scans/sec over a scanning range of 40–850 m/z. Raw mass spectral data were processed using the automated mass spectral deconvolution and identification system for peak deconvolution, while chromatographic analysis was conducted using GC-MS Solution software (Shimadzu Corp.). Compound identification was achieved by spectral matching against NIST8 and WILEY8 mass spectral libraries.
Metabolite quantification was based on triplicate measurements, with fold changes calculated relative to control samples. Peak areas were normalized against the internal standard (adonitol) and fresh plant weight, followed by logarithmic transformation for pseudo-quantification. Statistical analysis included univariate (two-sample t-test) and multivariate approaches (principal component analysis [PCA], partial least square discriminant analysis [PLS-DA]) using MetaboAnalyst 5.0 [22]. Missing values were imputed using k-nearest neighbor methodology, and data normalization employed Pareto scaling to enhance low-abundance metabolite detection while minimizing noise amplification. Metabolic pathway analysis was performed using MetPA in MetaboAnalyst to identify pathways affected by PGPB and AMF treatments through combined pathway enrichment and topology analysis. Common metabolites detected in all the samples were submitted to MetPA with annotation based on compound names, and the Kyoto Encyclopedia of Genes and Genomes library was selected in the analysis module. Pathway enrichment employed a hypergeometric distribution to calculate raw enrichment P-values, which were subsequently adjusted for multiple testing by Benjamini–Hochberg FDR. Pathways with Adj P < 0.05 were considered significant.
2.6. Molecular Docking Analysis of Differentially Expressed Proteins and Metabolites
The differentially expressed proteins in wheat grains of tripartite (CP4+AHP3+AMF) treatment identified through proteomics [Table S4 and 9] having log2foldchange ≥1 and P < 0.09 were selected for the protein-metabolite molecular docking analysis in AutoDock Vina. 70 proteins [Table S9] were used in docking against 30 metabolites (except carboxylic acid) upregulated in wheat grains, as shown in Figure 1a. The accessions of all proteins were searched against the UniProt database [23]. In cases where related homologues are present, the best protein candidate(s) were selected based on structural quality parameters and evidence of protein existence (protein level, transcript level, inferred from homology, or predicted). For example, P93692 was selected out of three accession numbers, H9AXB3, P93692, and A0A3B6KQL2, because P93692’s existence was previously confirmed at the protein level and, therefore, exhibited a higher annotation score. The three-dimensional (3D) structures of metabolites were accessed from the PubChem database [24] in Structure-Data File (SDF) format. The corresponding PubChem compound IDs for these metabolites are listed in Table S10. As the 3D conformation of stearic acid (CID 5281) was unavailable, the flat 2D SDF conformation was used to generate a 3D model by OpenBabel through energy-minimization in the PyRx desktop application [25]. The 3D structures of selected proteins [Table S9] were accessed from the Protein Data Bank, if an experimentally determined structure is available, or from the AlphaFold Protein Structure Database [26] in Protein Data Bank (PDB) format. AlphaFold is a highly accurate protein structure prediction tool demonstrating accuracy close to experimental structures, especially monomers [27]. Two quality control parameters, per-residue model confidence score and predicted aligned error provided along with all models generated by AlphaFold, were checked to confirm the quality of all protein models used in docking.
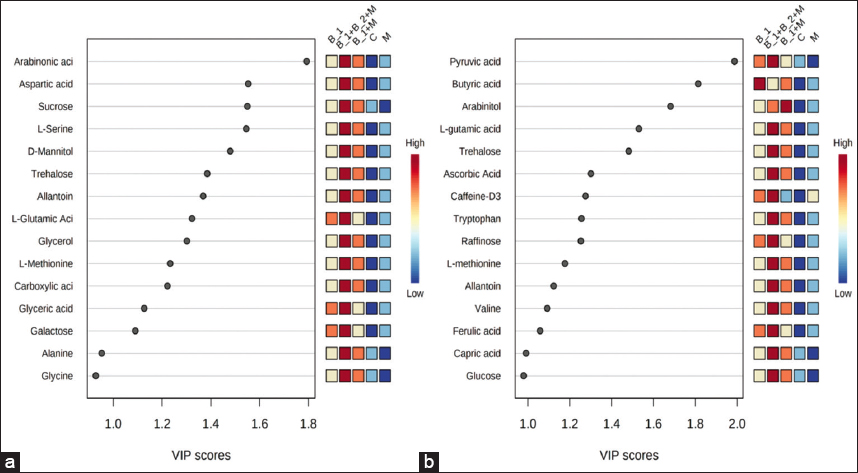 | Figure 1: Variable importance from validated PLS-DA models in MetaboAnalyst for wheat metabolites. Plots show metabolites ranked by variable importance in projection for (a) grain and (b) root tissues; adjacent miniheat blocks indicate relative abundance across treatment groups (B1 = CP4, B1+B2+M = CP4+AHP3+AMF, B1+M = CP4+AMF, C = control, M = AMF), with red and blue denoting higher and lower relative levels, respectively. AMF: Arbuscular mycorrhizal fungi, PLS-DA: Partial least squares discriminant analysis. [Click here to view] |
All molecular docking studies of the above proteins and metabolites were performed in the Ubuntu 22.04.4 operating system installed on Dell Optiplex-5050 hardware with Intel® Core™ i7-7700 CPU. AutoDock Vina docking program was used for molecular docking [28]. PyRx v0.9.2, a Python-based molecular screening software containing Vina as its docking program, was employed to prepare receptor and ligand input files for docking [25]. To get 3D structures with proper bond lengths between different atoms, all metabolite SDF files were energy-minimized using OpenBabel with default parameters in PyRx, i.e., universal force field method, and conjugate gradients as the optimization algorithm. Subsequently, the energy-minimized 3D conformations of metabolites were converted to the AutoDock ligand files in PDBQT format using AutoDock Tools in PyRx. Similarly, the protein PDB files were converted to PDBQT format in PyRx using the make macromolecule option to generate receptor files for AutoDock Vina. The Vina Search Space was maximized for all protein receptors, and docking was performed with default parameters (exhaustiveness = 8, energy range = 3).
The AutoDock Vina predicted ranked poses with associated binding affinity (ΔG) in Kcal/mol for each docking run. ΔG is the estimated binding free energy calculated by the scoring function from the docking algorithm. The protein-metabolite complexes with ΔG ≤−6.0 Kcal/mol were visualized in PyMOL v2.6.0, and the best-ranked conformation (root mean square deviation = 0) for the corresponding protein-metabolite complex was saved in PDB format.
LigPlot+ v.2.2.8 software was used to generate the 2D plots of protein–metabolite interactions using the complex PDB files as input and the allowed bond length parameters set to default. LigPlot calculates the hydrogen-bond (H-bond) distances between metabolite atoms and the protein residues’ atoms involved in H-bonding, along with all potential hydrophobic interactions in the binding pocket of a protein. The 2D diagram images generated by LigPlot in PostScript format were converted to PNG format.
3. RESULTS
In the current study, proteomic and metabolomic analyses of wheat (HD3086) treated with a microbial consortium of two PGPB (CP4, AHP3) and AMF and their different combinations were analyzed based on their superior performance in the 2nd year of field study [19]. In mature wheat grains, 88 proteins were significantly upregulated (log2fc >1, P < 0.05) in CP4+AHP3+AMF-inoculated plants, whereas 76 in CP4+AMF-inoculated, 49 in AMF-inoculated, and 98 proteins in CP4-inoculated plants were significantly upregulated compared to vehicle-treated (control) plants [Tables S1-4]. Similarly, 70, 43, 48, and 34 proteins were significantly downregulated in CP4-, AMF-, CP4+AMF-, and CP4+AHP3+AMF-inoculated plants, respectively, over control [Tables S5-8]. As many of these proteins are homologous, a comparative analysis of all the PGPB/AMF-treated samples revealed 9 unique proteins significantly upregulated and 2 unique proteins significantly downregulated in all four microbial treatments [Table 1].
Table 1: Differentially expressed (absolute log2foldchange ≥1) proteins in grain tissue of PGPB/AMF-inoculated wheat (HD3086) plants. Colored boxes indicate upregulation (red) and downregulation (blue) with comparison to the control. Statistical significance is denoted by *(P<0.05) and **(adj P<0.05, Benjamini–Hochberg FDR), n=2.
| Log2foldchange | ||||||
|---|---|---|---|---|---|---|
| Sr. No. | UniProt accession | Annotation | CP4 | AMF | CP4_AMF | CP4_AHP3_AMF |
| 1. | A0A3B6ST91 | Histone H2A (nucleosomal DNA-binding, protein heterodimerization activity) | 5.19** | 2.25* | 1.86* | 4.19** |
| 2. | A0A3B6KGS8 | ATP-dependent helicase domain-containing protein (5’- 3’ DNA helicase activity, P-loop containing nucleoside triphosphate hydrolase superfamily) | 1.33* | 1.59* | 5.19** | 3.98** |
| 3. | Q00445 | Small heat shock protein, chloroplastic, GN=HSP21 (response to stress) | 2.95** | 3.17** | 2.77** | 1.91* |
| 4. | Q8H0B8 | Cold-regulated protein GN=Wcor18 (phosphatidylethanolamine lipid-binding, serine protease inhibition, regulation of signaling pathways) | 2.35** | 3.01** | 2.1* | 2.6** |
| 5. | A0A3B6ETW5 | S-adenosylmethionine-dependent methyltransferase (methylation) | 0.04 | 4.92** | 1.87* | 2.94** |
| 6. | A0A3B6NTF1 | ATP synthase subunit O, mitochondrial (proton-transport, ATP synthesis) | 2.16 | 2.24* | 3.37** | 1.54 |
| 7. | A0A077RWR2 | SHSP domain-containing protein GN=TRAES_3BF066700050CFD_c1 (unfolded protein binding, response to hydrogen peroxide, heat, and salt stress) | 1.9** | 1.94* | 2.01* | 1.69* |
| 8. | A0A3B6SLX4 | 40S ribosomal protein S21 (endonucleolytic cleavage, translation) | 1.97** | 1.7* | 1.79* | 1.89* |
| 9. | A0A3B6ECS7 | PH domain-containing protein (ricin B-like lectin EULS3-like family, carbohydrate binding, stress-related euonymus-related lectins (EULs) in rice, Arabidopsis EULS3 involved in stomatal closure) | 1.51** | 1.86* | 1.92* | 1.96* |
| 10. | P93692 | Serpin-Z2B (serine-type endopeptidase inhibitor activity, extracellular space) | 1.51** | 1.15 | 2.95** | 1.49* |
| 11. | A0A3B6TQ10 | Bifunctional inhibitor/plant lipid transfer protein/seed storage helical domain-containing protein (non-specific lipid-transfer, proteinase/alpha-amylase inhibition, highly conserved cysteines) | 1.19* | 1.39* | 1.68* | 2.12* |
| 12. | D0EWS4 | Avenin-like b7 (nutrient reservoir activity, seed storage protein) | 1.01* | 1.49* | 2.32* | 1.54* |
| 13. | Q96123 | Superoxide dismutase [Cu-Zn] GN=SOD1.1 (antioxidant: removal of superoxide radicals, Cu-Zn SOD family, chloroplast) | 2.1** | 1.31 | 1.2 | 1.63* |
| 14. | A0A3B6MZC2 | H15 domain-containing protein (nucleosome assembly, chromosome condensation, double-stranded DNA binding, negative regulation of DNA recombination) | 1.33* | 1.48* | 1.18 | 1.52* |
| 15. | A0A3B6IQ78 | Gamma-interferon-inducible lysosomal thiol reductase (oxidoreductase, acting on sulfur group of donors, disulfide as acceptor) | 1.39* | 1.21 | 1.67* | 1.21* |
| 16. | A0A3B6ED63 | Why domain-containing protein (response to desiccation) | −2.08** | −2.23* | −1.81* | −0.82 |
| 17. | A0A3B6RIV9 | ADK_lid domain-containing protein (adenylate kinase, ATP-binding, transferase activity, biosynthesis) | −1.57** | −2.61* | −1.77* | −0.87 |
| 18. | Q2TN84 | USP family protein (Universal stress protein) | −1.12* | −2.06* | −1.39 | −2.25* |
| 19. | W5EH10 | Ribonucleoprotein (RNA-binding, mRNA splicing, box C/D methylation guide snoRNP complex) | −2.72** | −2.21* | −1.98* | −1.8* |
| 20. | A0A341P0J7 | Ribosomal_L18e/L15P domain-containing protein (mRNA-binding; translation) | −2.02** | −1.21 | −1.88* | −4.04** |
| 21. | A0A3B6FKN7 | Histone H2A/H2B/H3 domain-containing protein (structural constituent of chromatin, protein heterodimerization activity) | −2.09** | −3.21** | −3.24** | −1.72* |
| 22. | A0A3B5XY29 | O-acyltransferase (Glycerolipid metabolism, triacylglycerol biosynthesis) | −0.53 | −2.81* | −4.72** | −3.36** |
PGPB: Plant growth-promoting bacteria, FDR: False discovery rate
3.1. Wheat HD3086 Inoculation with PGPB and/or AMF Modulates different seed Storage and Defense-related Proteins
Seed storage proteins, namely Em protein, glutenin, gliadin, and avenin, were differentially expressed in PGPB/AMF-treated plants compared to control. The Em protein H5 (P42755) was downregulated 6.4-fold in CP4+AHP3+AMF-, 5.1-fold in CP4+AMF-, and 9-fold in CP4-inoculated plants over control. Similarly, Em proteins (P04568 and Q9ZR70) were also significantly downregulated in CP4+AMF-inoculated plants [Table S7]. The prolamin A0A3B6U5I1 was significantly upregulated (2.4-fold) while glutenin subunit protein (P16315) was significantly downregulated (2.1-fold) in CP4-inoculated plants compared to control [Tables S1 and S5]. The gamma gliadin-B2 (M9TLK0) was significantly upregulated 3.4-fold in CP4-, 2.7-fold in CP4+AHP3+AMF-, and 10-fold in CP4+AMF-treated plants [Table S1 and S3-4]. Alpha/beta gliadin (P04727) was downregulated 3.8-fold in CP4+AMF-inoculated plants [Table S7]. Gliadin/avenin-like seed protein (D2KFG9) was significantly upregulated (>2-fold) in CP4+AHP3+AMF- and AMF-treated plants over control [Table S2 and 4]. Avenin-like b7 protein (D0EWS4) was also upregulated by 2-fold in CP4-, 2.8-fold in AMF-, 2.9-fold in CP4+AHP3+AMF-, and 5-fold in CP4+AMF-treated plants [Table 1]. Similarly, multiple avenin-like proteins exhibited differential expression. Seed maturation protein (A0A3B6RKV2) involved in dormancy was significantly downregulated by 4.2-fold in CP4+AMF- treatment, whereas late embryogenesis abundant protein (A0A3B6ED63) was significantly downregulated in CP4-, AMF-, and CP4+AMF- treatments over control [Table 1 and S7]. Inositol-pentakisphosphate 2-kinase (A0A3B6J0F1), a key enzyme involved in phytate biosynthesis, was found significantly upregulated (2.5-fold) in AMF-inoculated wheat compared to control [Table S2].
In addition to seed storage proteins, multiple proteins associated with protection against phytopathogens were modulated in response to PGPB/AMF treatment compared to the control. Defensin protein A0A060APV2 was significantly downregulated (2-fold) in CP4-treated plants [Table S5], whereas defensin W5FHQ6 was significantly upregulated (2.8-fold) in CP4+AMF treatments [Table S3]. Similarly, defensin protein A0A060AQ78 showed significant upregulation in CP4+AMF- (3.4-fold), CP4+AHP3+AMF-, and AMF- (~2.3-fold) treated plants compared to control [Table S2-4]. WRKY are key proteins involved in defense regulation and innate immunity. WRKY protein A0A3B5ZXF4 was significantly upregulated in AMF-treated plants (5-fold) over control [Table S2]. Pathogenesis-related osmotin/thaumatin-like proteins (A0A3B6JDF8 and A0A3B6JER2) were significantly downregulated in CP4 treatments over control [Table S5]. Proteins involved in plant defense against insect or pathogen digestive enzymes (serine proteases, α-amylases) were differentially expressed. Dimeric α-amylase inhibitor Q4U1A4 was significantly upregulated (>2-fold) in AMF-, CP4+AMF-, and CP4+AHP3+AMF- treatments [Table S2-4], whereas α-amylase inhibitor 0.19 (P01085) was upregulated 2-fold (P = 0.089) in CP4+AHP3+AMF- inoculated plants compared to control. Cysteine proteinase inhibitor A0A3B6GQ71 was significantly upregulated (3.6-fold) in CP4+AHP3+AMF- treatments whereas significantly downregulated (2-fold) in CP4-treated wheat compared to control [Tables S4 and 5]. Similarly, Bowman-Birk type trypsin inhibitor P81713 was significantly upregulated in CP4+AMF- and CP4+AHP3+AMF-treated wheat compared to control [Tables S3 and 4].
3.2. Cellular Metabolism and Homeostasis-Related Proteins Modulated by PGPB/AMF in Wheat HD3086
Fructose-bisphosphate aldolase (A0A1D5VD11), a central enzyme in plant carbohydrate metabolism, was significantly downregulated in CP4+AMF- (5.7-fold) and AMF- (3.5-fold) inoculated wheat HD3086 compared to control [Tables S6 and 7]. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (W5I2D4), a key enzyme in glucose metabolism and maintenance of cellular adenosine triphosphate (ATP) levels, was significantly upregulated in CP4- (2.8-fold) treated plants compared to control [Table S1]. Starch synthase (Q9LEE2) expressed in plastids (amyloplast and chloroplast) was significantly upregulated in CP4+AMF- (5.2-fold) and CP4- (2.1-fold) treated plants compared to control [Table S1 and 3]. The endogenous alpha-amylase/subtilisin inhibitor (P16347) prevents the uncontrolled breakdown of starch in developing seeds. It was significantly upregulated in CP4- (2.2-fold) and CP4+AMF- (2.7-fold, P = 0.054) treated plants [Table S1]. Xylose isomerase (W5C4N4), involved in pentose sugar metabolism, was upregulated in CP4+AHP3+AMF- (1.96-fold, P = 0.098) treated plants compared to control. Adenylate kinase (A0A3B6RIV9), a central enzyme in rapid adenylate recycling, was significantly downregulated in CP4-, AMF-, and CP4+AMF- treatments over control [Table 1]. Alcohol dehydrogenase 1 (A9U8G4) was significantly upregulated in CP4- and tripartite treatments [Tables S1 and 3], whereas betaine-aldehyde dehydrogenase (A0A3B6PRJ5) and mitochondrial NADH dehydrogenase (ubiquinone) Fe-S protein 1 (A0A3B6HYA0) were significantly downregulated in AMF- treatments over control [Table S6]. Helicases function in DNA replication, transcription, and repair and are involved in maintaining the stability and integrity of the genome. Helicase A0A3B6KGS8 was highly upregulated in CP4+AMF- (~36-fold) and CP4+AHP3+AMF- (~15-fold) treatments, followed by AMF- (3-fold) and CP4- (2.5-fold) treatments compared to control. Similarly, histone proteins (A0A3B6ST91 and A0A3B6MZC2) were also significantly upregulated [Table 1].
Multiple redox homeostasis-related proteins were significantly modulated in PGPB- and AMF-treated plants. Superoxide dismutase [Cu-Zn] (Q96123), involved in the removal of superoxide radicals, was found significantly upregulated in CP4- (4.2-fold) and CP4+AHP3+AMF- (3.1-fold) treated plants compared to control [Table 1]. Peroxidase (A0A077S7C3) was significantly upregulated by ~3-fold in CP4+AMF-, AMF-, and CP4+AHP3+AMF-treated wheat plants over control [Tables S2-4]. L-ascorbate peroxidase (APX) (A0A3B6AR02), however, was significantly downregulated (3.7-fold) in AMF-treated wheat (HD3086) compared to control [Table S6]. Similarly, peroxiredoxin (A0A3B6ELY8) was significantly downregulated by 2.7-fold in CP4-treated samples compared to control [Table S5]. Apart from detoxification, peroxiredoxin family enzymes also regulate cellular redox signaling by sensing and responding to peroxide levels. Glutaredoxin (A0A2X0TY93), involved in cellular response to oxidative stress, was also significantly downregulated (2.9-fold) in CP4-treated plants over control [Table S5]. Gamma-interferon-inducible lysosomal thiol reductase (A0A3B6IQ78) was significantly upregulated in all microbial treatments over control [Table 1].
Several small heat shock proteins (sHSPs) were found to be differentially expressed in wheat HD3086 inoculated with PGP microbes compared to the control. A0A077RWR2 (cytoplasmic sHSP) and Q00445 (chloroplastic sHSP) were significantly upregulated in all microbial treatments [Table 1]. Similarly, chloroplast sHSP (Q9SBB7) was significantly upregulated (>2 fold) in CP4-, AMF-, and CP4+AMF-treated plants over control [Tables S1-3]. Another SHSP domain-containing protein (A0A3B6JJG2) was significantly upregulated (4.4-fold) in CP4+AMF-treated plants compared to control [Table S3].
3.3. Differentially Expressed Proteins with Uncharacterized Functions in PGPB/AMF-Treated Wheat HD3086
S-adenosylmethionine-dependent methyltransferase (A0A3B6ETW5) and O-acyltransferase (A0A3B5XY29) were significantly up- and downregulated, respectively, in AMF-, CP4+AMF-, and CP4+AHP3+AMF- treatments compared to control [Table 1]. USP family protein Q2TN84 was significantly downregulated in CP4-, AMF-, and CP4+AHP3+AMF-treated wheat compared to control [Tables S5, 6, 8]. Similarly, zinc finger protein D7NXU0 was significantly downregulated in CP4+AMF- treatment over control [Table S7]. Cold shock domain protein A0A3B5Y1S1 was significantly upregulated 3-fold in CP4- treatments over control [Table S1]. Cytochrome b5 heme-binding domain-containing protein (A0A3B6H417) was significantly upregulated in CP4 and CP4+AHP3+AMF treatments over control [Tables S1 and 4]. Uncharacterized seed protein A0A3B6TQ10, showing sequence homology with plant lipid-transfer proteins, also exhibited significant upregulation in all four microbial treatments, with maximum upregulation (4.3-fold) in CP4+AHP3+AMF-treated plants. Similarly, pleckstrin-homology (PH) domain-containing protein (A0A3B6ECS7) and cold-regulated protein (Q8H0B8) were significantly upregulated in wheat grains in all four microbial treatments compared to the control [Table 1]. Fasciclin-like protein FLA31 (Q06I75), a plant cell wall glycoprotein, was significantly upregulated (2.1-fold) in CP4-inoculated plants compared to control [Table S1]. Knot1 domain-containing protein (A0A3B5XUE9) was significantly upregulated in AMF-, CP4+AMF-, and CP4+AHP3+AMF- treatments [Tables S2-5], whereas another knottin (A0A3B6IYR0) was significantly downregulated in CP4-treated wheat over control [Table S5]. Uncharacterized family protein (A0A3B6U6Y8) containing Bet v I/Major latex protein domain was significantly upregulated in CP4+AMF- and CP4+AHP3+AMF- treatments over control [Tables S3 and 4]. Plant dehydrin family protein Rab (Q41579) was significantly upregulated in CP4- and CP4+AHP3+AMF- treatments compared with control [Tables S1 and 4].
3.4. CP4+AHP3+AMF-Treated Wheat Exhibits Differential Expression of Novel Proteins
In tripartite (CP4+AHP3+AMF) microbial consortia (not tested in 1st-year field trial)-treated wheat (HD3086), several proteins were found to be significantly differentially expressed [Tables S4 and 8]. Clathrin protein (A0A3B6B9R9) was significantly downregulated (3.2-fold) only in CP4+AHP3+AMF-inoculated plants compared to the control. Clathrins are involved in endocytosis and intracellular protein transport. Subtilisin-chymotrypsin inhibitor-2A (W5D700) was significantly downregulated (3.5-fold) in tripartite treatment over control. Similarly, uncharacterized protein A0A3B6PPD5 (homologous to NFU1 iron-sulfur cluster protein M1B4H6 in S. tuberosum) was significantly downregulated by 3.4-fold in CP4+AHP3+AMF-treated samples over control [Table S8]. EC protein I/II (P30569), potentially involved in zinc ion homeostasis, was significantly upregulated (2.7-fold) in CP4+AHP3+AMF-treatment compared to control. Plasma membrane ATPase (A0A3B6JMY4), involved in intracellular pH regulation, was significantly upregulated (9-fold) in only tripartite treatments over control. Similarly, gamma-interferon-inducible lysosomal thiol reductase (A0A3B6JLE2), Bowman-Birk serine protease inhibitor (Q6QAX7), and bifunctional inhibitor/plant lipid transfer protein (LTP)/seed storage helical domain-containing proteins (A0A3B5XVX5 and A0A3B6KQR9) were significantly upregulated specifically in CP4+AHP3+AMF-inoculated wheat plants compared to control [Table S4].
3.5. Inoculation with PGPB and AMF Significantly Altered Metabolites in Grain and Root Tissues of Wheat HD3086
Metabolomics of wheat grain and root tissues at the maturity stage revealed significant changes in PGPB/AMF-inoculated plants compared to uninoculated controls. Supervised analysis was performed in MetaboAnalyst to complement the unsupervised PCA [Figure S7] which shows gross changes in metabolomic profiling in the grain and root by variance explained by principal components.PLS-DA models were trained with K-fold cross-validation to select components and estimate predictivity. The retained model achieved R2Y and Q2 indicative of good fit and generalization whereas permutation testing confirmed that observed Q2 exceeded the null distribution thus supporting non-random class separation. The top discriminating metabolites in grain and root tissues were identified based on variable importance in projection (VIP) score >1 and P < 0.05. A total of 13 metabolites in grain and 14 metabolites in root were determined as potential markers [Figure 1a and b]. L-glutamic acid, trehalose, allantoin, and L-methionine exhibited significantly high VIP scores in both grain and root tissues. Prominent metabolites in grain include arabinonic acid, aspartic acid, and sucrose whereas root profiles showed pyruvic acid, butyric acid, and arabinitol among the top contributors in the discriminant analysis.
The differential expression analysis of metabolites in PGPB/AMF-treated wheat revealed 31 and 35 metabolites significantly modulated in grain and root tissues, respectively, relative to uninoculated control [Figure 2]. In wheat grains, palmitic acid, inositol, sucrose, maltose, trehalose, allantoin, aspartic acid, and oleic acid were elevated ~2-fold in CP4+AHP3+AMF- treatments relative to control. The CP4+AMF- and CP4+AHP3+AMF- treatments exhibited several metabolites that were shared upregulated, including alanine, xylitol, caprylic acid, stearic acid, valine, jasmonic acid, succinic acid, methionine, mannitol, and serine. The coordinated increases in amino acids, sugars, polyols, and medium-chain and long-chain fatty acids in consortia treatments over uninoculated control indicate a concerted shift in carbon-nitrogen metabolism and lipidomic profile, particularly under tripartite treatment. Pyruvic acid showed treatment-specific behavior, remaining elevated in CP4- and AMF- treatments relative to control but less pronounced in consortia treatments, suggesting that the combined treatments channel carbon beyond glycolytic endpoints toward downstream storage compounds. In CP4- and AMF- treatments, jasmonic acid and maltose exhibited regulation in opposite directions, indicating that CP4 and AMF elicit distinct signaling and carbohydrate responses in grain [Figure 2a]. Hierarchical clustering grouped CP4+AHP3+AMF replicates into a distinct branch separated from control and single inoculations, with most of the grain metabolites peaking in this treatment [Figure 2a]. It is consistent with the PCA exploratory structure [Figure S7] and with VIP-highlighted sugars and amino acids contributing to supervised discrimination [Figure 1].
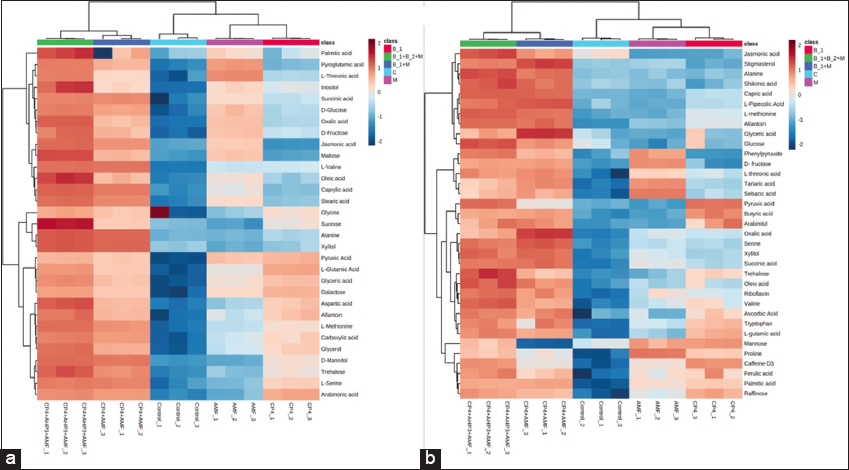 | Figure 2: Log2 foldchange heatmaps with hierarchical clustering of metabolites across treatments in wheat HD3086 (a) grain and (b) root tissue. Colors denote relative increase (red) or decrease (blue) versus control, and dendrograms indicate metabolite clustering patterns. Treatment labels: B1 = CP4, B1+M = CP4+AMF, B1+B2+M = CP4+AHP3+AMF, M = AMF, C = control. AMF: Arbuscular mycorrhizal fungi. [Click here to view] |
The consortia treatments also led to significant upregulation of wheat root metabolites compared to control [Figure 2b]. Tripartite inoculation of wheat exhibited >2-fold increases in jasmonic acid, alanine, shikimic acid, capric acid, glucose, phenylpyruvate, pyruvic acid, tryptophan, trehalose, and oleic acid, indicating coordinated activation of carbon flux, aromatic biosynthesis, and signaling pathways. Similarly, bipartite inoculation exhibited marked increases (>2-fold) in stigmasterol, shikimic acid, jasmonic acid, capric acid, glyceric acid, and oxalic acid over the control. Proline expression was lower in bipartite yet higher in other inoculations compared to the control, indicating treatment-specific amino acid regulation patterns. Single inoculants, bacterial (CP4) and fungal (AMF), showed narrower responses instead. CP4 increased pyruvic and butyric acids, arabinitol, mannose, caffeine, and raffinose, while AMF elevated phenylpyruvate, fructose, tartaric and sebacic acids, mannose, and proline over control. CP4- and AMF- treatments exhibited significant downregulation in jasmonic acid, however. The dendrogram splits in Figure 2b reflect these contrasting patterns. The root metabolome was altered significantly under microbial treatments, with combined PGPB-AMF inoculations jointly enhancing central carbon intermediates, aromatic pathway precursors, and membrane or stress-associated lipids.
The correlation heatmaps revealed coherent metabolite modules in both the tissues [Figure 3]. The prominent positively correlated block linking sugars (sucrose, maltose, and glucose), polyols (mannitol, arabinitol, and xylitol), and amino acids (alanine, serine, methionine, and valine) is consistent with the high-intensity clusters in the differential abundance heatmaps and VIP-ranked metabolites such as trehalose, L-methionine, pyruvate, and ascorbate [Figures 1 and 2]. Distinct correlation groups among organic and fatty acids (oxalic acid, glyceric acid, capric acid, stearic acid, and oleic acid) mirror treatment-specific abundance patterns, therefore, supporting the companion analyses and indicating that combined inoculations modulate interconnected metabolic pathways rather than isolated metabolites.
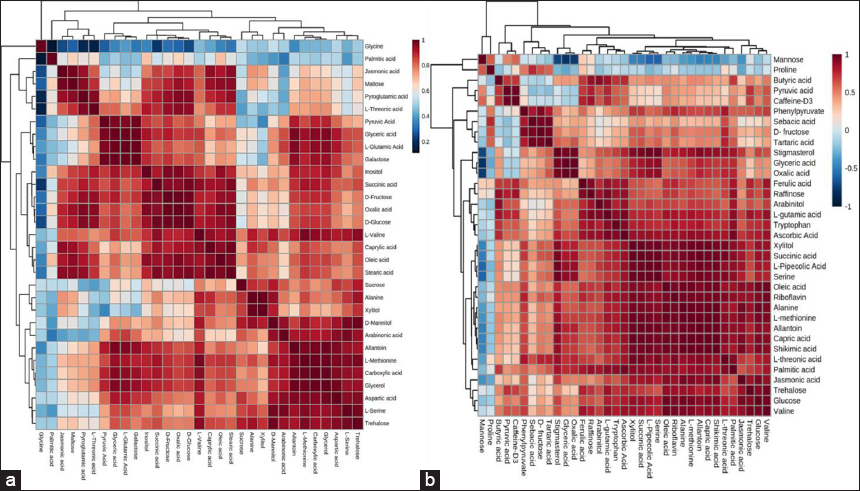 | Figure 3: Correlation analyses of differentially expressed metabolites represented by heatmaps in wheat HD3086 (a) grain and (b) root tissue. Colors indicate strength and direction of pairwise correlations and hierarchical clustering group metabolites with similar co-variation patterns. [Click here to view] |
3.6. Metabolic Pathway Analysis Identified Diverse Upregulated Pathways
MetPA identified 29 unique pathways returned by enrichment analysis [Tables 2 and 3]. Relevant metabolic pathways modulated in wheat tissues were identified based on -log(P-value) and pathway impact scores. Topology-based impact score measures the centrality of significant metabolites identified in a pathway. Metabolic pathway analysis in wheat grain identified five pathways that were statistically significant after multipletesting correction (FDR <0.05), namely glyoxylate and dicarboxylate metabolism (impact 0.22), aminoacyltRNA biosynthesis (impact 0.11), alanine, aspartate and glutamate metabolism (impact 0.45), glycine, serine and threonine metabolism (impact 0.42), and galactose metabolism (impact 0.042). Additional pathways showed nominal enrichment at raw P < 0.05, including carbon fixation in photosynthetic organisms (impact 0.04), cysteine and methionine metabolism (impact 0.14), glutathione metabolism (impact 0.13), fatty acid biosynthesis (impact 0.01), and cutin, suberine, and wax biosynthesis (impact 0.12) [Table 2]. MetPA analysis in wheat roots returned all pathways with adjusted P ≥ 0.205; however, several pathways exhibited nominal enrichment at raw P < 0.05. These metabolic pathways include aminoacyl-tRNA biosynthesis, butanoate metabolism, alanine, aspartate and glutamate metabolism, and phenylalanine, tyrosine, and tryptophan biosynthesis. Additional pathways with weaker nominal signals included glyoxylate and dicarboxylate metabolism (impact 0.045) and glycine, serine, and threonine metabolism (impact 0.02) [Table 3]. Comparative pathway analysis between grain and root showed that 11 pathways were common in response to PGPB- and AMF- treatment.
Table 2: Pathway analysis in wheat grain tissue using KEGG.
| Pathway name | Match status | P | -log (p) | Holm p | FDR | Impact |
|---|---|---|---|---|---|---|
| Glyoxylate and dicarboxylate metabolism | 6/29 | 1.3953E−5 | 4.8553 | 0.0013395 | 9.884E−4 | 0.21749 |
| Aminoacyl-tRNA biosynthesis | 7/46 | 2.0592E−5 | 4.6863 | 0.0019562 | 9.884E−4 | 0.11111 |
| Alanine, aspartate, and glutamate metabolism | 5/22 | 4.9638E−5 | 4.3042 | 0.0046659 | 0.0015884 | 0.44964 |
| Glycine, serine, and threonine metabolism | 5/33 | 3.8249E−4 | 3.4174 | 0.035571 | 0.0091797 | 0.41558 |
| Galactose metabolism | 4/27 | 0.0017303 | 2.7619 | 0.15919 | 0.033222 | 0.04252 |
| Butanoate metabolism | 3/17 | 0.0041985 | 2.3769 | 0.38207 | 0.067177 | 0.0 |
| Carbon fixation in photosynthetic organisms | 3/21 | 0.0077744 | 2.1093 | 0.69969 | 0.10656 | 0.03607 |
| Biosynthesis of unsaturated fatty acids | 3/22 | 0.0088797 | 2.0516 | 0.79029 | 0.10656 | 0.0 |
| Monobactam biosynthesis | 2/8 | 0.010274 | 1.9883 | 0.90409 | 0.10959 | 0.0 |
| Cysteine and methionine metabolism | 4/46 | 0.012302 | 1.91 | 1.0 | 0.1181 | 0.13971 |
| Glutathione metabolism | 3/26 | 0.014195 | 1.8479 | 1.0 | 0.12388 | 0.13362 |
| Cyanoamino acid metabolism | 3/29 | 0.01915 | 1.7178 | 1.0 | 0.1532 | 0.0 |
| Fatty acid biosynthesis | 4/56 | 0.024048 | 1.6189 | 1.0 | 0.17759 | 0.01123 |
| Sulfur metabolism | 2/15 | 0.035285 | 1.4524 | 1.0 | 0.24195 | 0.03315 |
| Arginine biosynthesis | 2/18 | 0.049526 | 1.3052 | 1.0 | 0.29716 | 0.08544 |
| Cutin, suberine, and wax biosynthesis | 2/18 | 0.049526 | 1.3052 | 1.0 | 0.29716 | 0.125 |
| Citrate cycle (TCA cycle) | 2/20 | 0.059991 | 1.2219 | 1.0 | 0.32525 | 0.0401 |
| Glycerolipid metabolism | 2/21 | 0.065488 | 1.1838 | 1.0 | 0.32525 | 0.01703 |
| Thiamine metabolism | 2/22 | 0.071148 | 1.1478 | 1.0 | 0.32525 | 0.0 |
| Starch and sucrose metabolism | 2/22 | 0.071148 | 1.1478 | 1.0 | 0.32525 | 0.09857 |
KEGG: Kyoto encyclopedia of genes and genomes, FDR: False discovery rate, TCA: Tricarboxylic acid cycle
Table 3: Pathway analysis in wheat root tissue using KEGG.
| Pathway name | Match status | P | -log (p) | Holm p | FDR | Impact |
|---|---|---|---|---|---|---|
| Aminoacyl-tRNA biosynthesis | 5/46 | 0.0021377 | 2.67 | 0.20522 | 0.20522 | 0.0 |
| Butanoate metabolism | 3/17 | 0.0046154 | 2.3358 | 0.43846 | 0.22154 | 0.0 |
| Alanine, aspartate, and glutamate metabolism | 3/22 | 0.0097368 | 2.0116 | 0.91526 | 0.23368 | 0.0 |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | 3/22 | 0.0097368 | 2.0116 | 0.91526 | 0.23368 | 0.08859 |
| Glyoxylate and dicarboxylate metabolism | 3/29 | 0.020926 | 1.6793 | 1.0 | 0.40177 | 0.04494 |
| Glycine, serine, and threonine metabolism | 3/33 | 0.029532 | 1.5297 | 1.0 | 0.47251 | 0.02252 |
| Cutin, suberine, and wax biosynthesis | 2/18 | 0.052565 | 1.2793 | 1.0 | 0.60193 | 0.125 |
| Citrate cycle (TCA cycle) | 2/20 | 0.063617 | 1.1964 | 1.0 | 0.60193 | 0.0401 |
| Carbon fixation in photosynthetic organisms | 2/21 | 0.069416 | 1.1585 | 1.0 | 0.60193 | 0.03607 |
| Biosynthesis of unsaturated fatty acids | 2/22 | 0.075384 | 1.1227 | 1.0 | 0.60193 | 0.0 |
| Valine, leucine, and isoleucine biosynthesis | 2/22 | 0.075384 | 1.1227 | 1.0 | 0.60193 | 0.10727 |
| Indole alkaloid biosynthesis | 1/4 | 0.081108 | 1.0909 | 1.0 | 0.60193 | 0.0 |
| Pantothenate and CoA biosynthesis | 2/23 | 0.081512 | 1.0888 | 1.0 | 0.60193 | 0.0 |
| Glycolysis/gluconeogenesis | 2/26 | 0.10077 | 0.99667 | 1.0 | 0.69099 | 0.12074 |
| Fatty acid biosynthesis | 3/56 | 0.10879 | 0.96341 | 1.0 | 0.69626 | 0.01123 |
| C5-Branched dibasic acid metabolism | 1/6 | 0.11924 | 0.92359 | 1.0 | 0.71543 | 0.0 |
| Glucosinolate biosynthesis | 3/65 | 0.15138 | 0.81994 | 1.0 | 0.78737 | 0.0 |
| Monobactam biosynthesis | 1/8 | 0.15583 | 0.80734 | 1.0 | 0.78737 | 0.0 |
| Tropane, piperidine, and pyridine alkaloid biosynthesis | 1/8 | 0.15583 | 0.80734 | 1.0 | 0.78737 | 0.0 |
| Riboflavin metabolism | 1/11 | 0.20798 | 0.68197 | 1.0 | 0.95078 | 0.11852 |
KEGG: Kyoto encyclopedia of genes and genomes, FDR: False discovery rate, TCA: Tricarboxylic acid cycle
3.7. Protein–Metabolite Interaction Analysis using AutoDock Vina
The molecular docking analysis of significantly upregulated (log2fc ≥1) grain proteins in CP4+AHP3+AMF-treated wheat with upregulated grain metabolites in AutoDock Vina revealed ATP-dependent helicase domain-containing protein A0A3B6LIU4–Sucrose complex exhibited maximum binding affinity (−7.8 Kcal/mol), whereas Rab protein Q41579–Palmitic acid showed the weakest interaction (−2.2 Kcal/mol) [Figures 4d and S8]. Multiple significantly upregulated proteins-SHSP domain-containing protein A0A077RWR2, bifunctional inhibitor/plant LTP/seed storage helical domain-containing protein A0A3B6TQ10, superoxide dismutase O24400, cysteine proteinase inhibitor Q1XHC6, cytochrome b5 heme-binding domain-containing protein A0A3B6H417, EC protein I/II P30569, γ gliadin-B2 M9TLK0, and histones (A0A3B6ST91 and A0A3B6LV22) did not show any significant interaction with the upregulated metabolites.
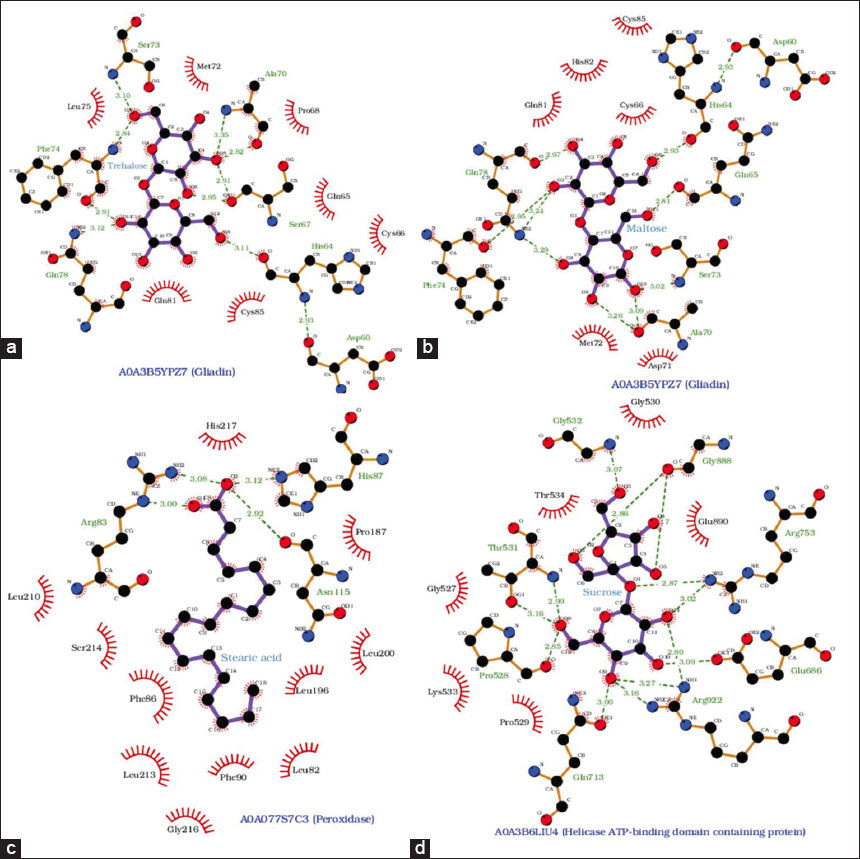 | Figure 4: LigPlot representation of hydrogen bonds and hydrophobic contacts between selected proteins and metabolites upregulated in CP4+AHP3+AMF-treated wheat HD3086. Seed storage protein–gliadin (A0A3B5YPZ7) interaction with (a) Trehalose, and (b) Maltose. (c) Peroxidase (A0A077S7C3) interaction with stearic acid, and (d) ATP-dependent helicase domain-containing protein (A0A3B6LIU4) interaction with sucrose. All interactions were predicted in molecular docking analysis by AutoDock Vina. Dashed green lines indicate hydrogen bonds with their predicted length in angstroms. [Click here to view] |
Prolamins (gliadins in wheat) are part of the alcohol-soluble fraction of gluten proteins. Six seed storage prolamins (A0A0K2QJB4, A0A3B5YPZ7, A0A3B6PHC0, D2KFG9, D0EWS4, and D6QZM4) with nutrient reservoir activity showed significant ΔG with sugar metabolites – trehalose, sucrose, maltose, and D-fructose [Table S11]. For example, the A0A3B5YPZ7 protein binds trehalose and maltose in a similar fashion with comparable binding energies, −7.0 Kcal/mol and −6.7 Kcal/mol, respectively. As evident from the LigPlot analysis of these complexes in Figure 4a and b, these disaccharides appear to bind A0A3B5YPZ7 in the same pocket region, as many interacting residues are conserved. While binding with trehalose and maltose, protein residues Ser73, Phe74, Gln78, His64, and Asp60 form H-bonds, whereas Met72, Cys66, Glu81, and Cys85 are involved in non-bonded hydrophobic contacts.
A0A077S7C3, a peroxidase, binds to stearic acid and allantoin with binding affinities of −6.8 Kcal/mol [Figure 4c] and −6.4 Kcal/mol, respectively [Table S11]. Apart from nitrogen mobilization through the xylem, allantoin’s role in abiotic stress tolerance is increasingly appreciated. Higher biosynthesis and accumulation of allantoin subsequently activate abscisic acid biosynthetic genes, which in turn activate hallmark downstream stress-related genes [29]. Allantoin also binds to Q4U1A4, a dimeric alpha-amylase inhibitor (AAI), and A0A3B6JQ63, a plasma membrane ATPase with −6.3 Kcal/mol ΔG [Figure S1a and 1b].
Several proteinase inhibitors of the bifunctional inhibitor/plant LTP/seed storage helical domain superfamily and metabolites bind with high affinity during molecular docking. A0A3B6MXN3 binds with jasmonic acid with a high affinity of −7.2 Kcal/mol, forming two H-bonds by O1 and O3 atoms of jasmonic acid with Arg27 and Met30 residues, respectively, whereas twelve other residues (Ile31, Cys34, Gly35, Thr38, Trp56, Ile61, Leu64, Leu68, Cys65, Leu68, Trp76, and Cys98) stabilize the complex through hydrophobic interactions [Figure S2a]. AAI domain-containing proteins, A0A3B6FHE6, A0A3B6GR96, and W5D003, showed good binding affinity toward sugar metabolites. A0A3B6GR96-trehalose complex exhibited −6.4 Kcal/mol binding affinity where Trp34, Cys36, Trp81, Val134, Val135, and Asp147 form H-bonds with the trehalose ligand, and Met35, Cys84, Gly85, Tyr88 residues form hydrophobic contacts [Figure S2b]. Another protein of this superfamily, A0A3B6KQR9, with only 62 amino acids (aa) chain length, binds oleic acid with −5.8 Kcal/mol ΔG. The complex is majorly stabilized by hydrophobic interactions, while Leu7 is the only residue forming an H-bond with O1 of oleic acid, which is the most widely distributed natural fatty acid [Figure S2c]. A0A3B6KQR9 may function as an LTP, an important family of proteins playing diverse physiological roles in land plants [30].
Other protease inhibitor proteins, including AAI (P01085 and Q4U1A4) and trypsin inhibitor (P81713 and P93692), also bind with maltose, trehalose, and sucrose with good binding energies ranging from −6.1 Kcal/mol to −6.8 Kcal/mol [Table S11]. Serpin-Z2B (P93692) is a serine-protease inhibitor belonging to the plant serpin superfamily involved in protease regulation in plants. Dimeric AAI Q4U1A4 binds inositol with a binding affinity of −6.1 Kcal/mol. A0A3B6A2J7, an aspartic-type endopeptidase related to the lipid-protein metabolic process, binds maltose disaccharide with a high affinity of −7.2 Kcal/mol. The complex shows extensive H-bonding and hydrophobic interactions [Figure S2d].
A0A3B6MUH5, a small cysteine-rich protein (80 aa) of knottin scorpion toxin-like superfamily and related to the plant’s defense response, binds trehalose with an affinity of −6.3 Kcal/mol [Figure S3a]. Defensins are integral components of the plant’s innate immune system. Another wheat defensin protein, A0A060AQ78 of the same length, encoded by the PDF16 gene, also binds trehalose but with a lower affinity of −5.7 Kcal/mol. Similarly, A0A3B6LF34, a thaumatin-like defense protein related to stress response, binds sucrose with −6.2 Kcal/mol affinity.
A0A3B6IQ78 protein of γ-interferon-inducible lysosomal thiol reductase family binds trehalose sugar with −6.1 Kcal/mol ΔG [Table S11]. Oxidoreductase activity of these lysosomal proteins catalyzes the disulfide bond reduction of their targets. A0A3B6ETW5, an uncharacterized protein of the S-adenosyl-L-methionine-dependent methyltransferase family, binds maltose with −6.4 Kcal/mol affinity. However, it also binds other disaccharides (sucrose and trehalose) and jasmonic acid in the same binding pocket but with comparatively lower affinity [Figure S3b].
A0A3B6KIU4, an uncharacterized protein of the At5g01610-like superfamily, binds caprylic acid with a good binding affinity of −6.3 Kcal/mol. The binding is supported by two H-bonds formed by Asp131 and His133 residues with the carboxyl group of caprylic acid, whereas twelve other residues form hydrophobic interactions in the binding pocket [Figure S3c]. A0A3B6PKG6, a membrane protein, binds maltose with −6.1 Kcal/mol ΔG [Figure S3d]. A0A3B6PKG6 contains a structural domain of six alpha-helices found in mitochondrial carrier proteins. It is involved in transmembrane organophosphate ester transport, and the sequence-similarity search revealed D0EY60, an ADP-glucose brittle-1 transporter, as its homolog in wheat with >90% identity.
A0A3B6NTF1, a component protein of the catalytic core (F1) of mitochondrial proton-transporting ATP synthase complex, binds maltose with −6.0 Kcal/mol ΔG. Proton-exporting P-type ATPase transporter proteins, A0A3B6JMY4 and A0A3B6JQ63, bind disaccharides (sucrose, maltose, and trehalose) with comparable binding free energies, −6.1 Kcal/mol to −6.5 Kcal/mol [Table S11]. A0A3B6JMY4 also binds jasmonic acid in its transmembrane helical domain with −6.2 Kcal/mol affinity [Figure S4a]. Plasma membrane P-type ATPases pump ions across the membrane against a concentration gradient, utilizing energy generated from ATP hydrolysis.
A cold-regulated protein Q8H0B8, encoded by the Wcor18 gene, binds sucrose and jasmonic acid with −5.9 Kcal/mol and −5.8 Kcal/mol ΔG, respectively [Figure S4b]. It shares 100% identity with A0A452ZBP4, a phosphatidylethanolamine-binding protein (PEBP) of goat grass, Aegilops tauschii. Proteins of the PEBP superfamily are involved in various functions, including lipid binding, serine protease inhibition, and switching between shoot growth and flower structures. A0A3B6ECS7, an uncharacterized protein containing a PH domain, binds maltose with −6.1 Kcal/mol ΔG [Table S11]. It shares homology with the ricin B-like lectin superfamily, whereas the ricin B lectin domain interacts with simple sugars. A0A3B6LW98 rRNA N-glycosylase, a plant defense protein with a toxic activity that leads to inhibition of translation, binds sucrose and maltose with −6.9 Kcal/mol and −6.8 Kcal/mol ΔG, respectively [Figure S5]. W5EAP3, a ribosomal biogenesis protein of the uL1 family and also a major structural constituent of the large subunit, binds trehalose with −6.2 Kcal/mol ΔG [Table S11].
Several highly upregulated ATP-binding helicase proteins also exhibited varying affinities toward disaccharides and other upregulated metabolites [Table S11 and Figure S6]. Allantoin binds A0A3B6KGS8 with −6.3 Kcal/mol ΔG. D-mannitol, a sugar alcohol implicated in antioxidant and reactive oxygen species (ROS) scavenging activities [31], binds A0A3B6KHG3 with −6.0 Kcal/mol ΔG [Figure S6a]. Arabinonic acid binds A0A3B6LKH3 with −6.0 Kcal/mol ΔG [Figure S6b]. Among all six helicases, molecular docking analysis predicted the highest affinity for A0A3B6LIU4 against sucrose (−7.8 Kcal/mol) and maltose (−7.7 Kcal/mol) [Table S11]. Stigmasterol, which was upregulated in root tissue of CP4+AMF- and CP4+AHP3+AMF- inoculated wheat plants [Figure 1b], showed high binding affinity (−8.7 Kcal/mol) with helicase protein A0A3B6KGS8 [Figure S8]. Stigmasterol is a structural component of plant cell membranes and, therefore, is also present in the shoot part of the plants.
4. DISCUSSION
The integration of proteomics and metabolomics revealed coordinated molecular mechanisms underlying PGPB- (CP4, AHP3) and AMF-mediated growth enhancement in wheat HD3086. These mechanisms comprise three interconnected biochemical systems: (1) Nutrient acquisition and biofortification, (2) energy metabolism modulation, and (3) stress tolerance through redox homeostasis. The upregulation of 88 proteins in tripartite (CP4+AHP3+AMF) treatment, including seed storage proteins (avenins, gliadins), stress-response proteins (heat shock proteins, defensins), energy-production enzymes (ATP synthases), and nucleic acid-processing enzymes (helicases), indicates comprehensive metabolic reprogramming [32]. These changes, coupled with the accumulation of 31 and 35 significantly altered metabolites in grain and root tissues, respectively, provide molecular insights into the enhanced growth, yield, and nutrient biofortification as observed under field conditions [19]. Tripartite treatment led to differential expression of some novel proteins, including clathrins, EC protein I/II, subtilisin chymotrypsin inhibitor 2A, and plasma membrane ATPase, suggesting that the addition of Bacillus sp. AHP3 in microbial consortia may elicit differential proteomic and metabolic responses in wheat HD3086 [Tables S4 and 8].
As the samples were collected at the harvesting (maturity) stage, the proteomics essentially captured the final nutrient allocation and grain quality. PGPB/AMF inoculation reprograms wheat seed-storage protein composition as evident in downregulated Em, glutenin, α/β-gliadin, and upregulated γ-gliadin, avenin-like, and plant lipid transfer-like proteins. The resultant shift in seed prolamin profile may alter the nutritional and processing quality of wheat HD3086. These changes are consistent with previous reports that microbial inoculation modulates the protein composition of wheat grain [33,34]. This protein remodeling aligns with the primary PGPB/AMF mechanisms of nutrient mobilization and availability.
Second, altered energy metabolism emerged as a coordinated primary mechanism. Downregulation of glycolytic enzyme [fructose-bisphosphate aldolase A0A1D5VD11; Tables S6 and 7] with concomitant upregulation of starch synthase [Q9LEE2; Table S1 and 3] and starch-protecting inhibitors [P16347; Table S1, and results section] indicates a shift in carbohydrate partitioning toward starch synthesis and protection during seed fill. GAPDH upregulation in CP4-treated plants suggests treatment-specific modulation of glycolytic and NAD+/ATP balance [31]. Downregulation of adenylate kinase [A0A3B6RIV9; Table 1] reflects a shift from active ATP turnover toward stable storage of carbon (starch) and lowered cytosolic ATP flux and AMP-mediated energy-stress signaling. Phytate is the main phosphorus storage molecule in cereals. AMF improves phosphorus uptake, and the plant likely channels this into phytate accumulation for storage in seeds, as evidenced by upregulated inositol-pentakisphosphate 2-kinase [A0A3B6J0F1; Table S2]. The concurrent upregulation of S-adenosylmethionine-dependent methyltransferase [A0A3B6ETW5; Table 1] and downregulation of O-acyltransferase [A0A3B5XY29; Table 1] in AMF- and AMF-inclusive consortia suggest a biochemical shift from acylation to methylation pathways.
The proteomic data show clear modulation of multiple pathogen-defence wheat proteins in response to PGPB/AMF inoculation. Downregulation of defensin (A0A060APV2) in CP4- treatments but upregulation of other defensins (e.g., W5FHQ6, A0A060AQ78) in AMF-containing treatments suggests differential defense regulation. PGPB alone may transiently reduce certain basal defenses, but AMF- or combined consortia strongly induce defensin-mediated antimicrobial protection. Defensin (antimicrobial peptides) can also play a role in seed storability by providing seed protection against storage fungi. The upregulation of WRKY transcription factor (A0A3B5ZXF4) in AMF-treatments suggests systemic priming of immune pathways maintained in mature seeds that may influence defense priming during germination of the offspring [35]. The significant upregulation of serpins [Serpin-Z2B P93692; Table 1] across multiple treatments suggests enhanced protease regulation which is crucial for seed protein stability and quality [33]. It is supported by amino acid preservation [alanine, aspartic acid; Figure 1 and 2] and correlates with field-observed grain quality improvement. Changes in these digestive enzyme inhibitors can also affect nutritional digestibility or allergenicity in humans.
Third, integrated stress tolerance through redox homeostasis is also evident. ROS accumulates during desiccation and storage or even during higher metabolic activity. The significant upregulation of superoxide dismutase (SOD) and peroxidase, along with strongly upregulated sHSPs across treatments [Table 1 and S1-4] and accumulation of antioxidant metabolites [allantoin, ascorbate; Figure 1], suggests that PGPB/AMF treatment induces a protective state preventing oxidative damage during late seed maturation. Downregulation of APX (in AMF-) as well as thiol-based redox regulators – peroxiredoxin (Prx) and glutaredoxin (in CP4-), however, indicates complexity in remodeling of redox homeostasis [36]. Higher SOD/peroxidase activity possibly improves seed longevity and vigor. Similarly, sHSPs act as molecular chaperones, improving seed proteostasis and membrane stability during seed desiccation [37]. Downregulation of APX and Prx in some treatments suggests a possible shift in redox management strategy that relies more on SOD/peroxidase pathways than APX/Prx-based fine-tuning. It implies that bulk ROS detoxification capacity is increased while sensitivity to redox signals might be reduced.
Apart from defence priming in seeds through defensins and inhibitors, as well as induced stress-tolerance through ROS enzymes and sHSPs, PGPB/AMF inoculation also likely induced chromatin remodeling in wheat seeds. The high co-induction of histone H2A (A0A3B6ST91) and helicase (A0A3B6KGS8) in all treatments [Table 1] suggests a coordinated program to protect the genome from oxidative or desiccation-induced damage in mature seeds. H2A components of nucleosome get phosphorylated at DNA double-strand breaks and recruit repair machinery, whereas helicases maintain genome stability during replication and repair. Higher abundance of these proteins might be associated with transcriptional reprogramming and stress-memory imprinting in the seeds [38]. CP4-treated wheat exhibited the strongest H2A upregulation [36-fold; Table 1] but weaker defense and ROS detoxification [e.g., lower defensin and glutaredoxin; Table S5]. This may reflect a compensatory mechanism for genome stability in the absence of strong AMF-mediated stress buffering. Treatments containing AMF show balanced upregulation of H2A (3-18-fold) coupled with better ROS control, chaperone activity, and defense priming. This suggests that AMF moderates the need for extreme chromatin restructuring by supporting other protective pathways.
The metabolomic analysis revealed coordinated changes in primary and secondary metabolism that support the observed proteomic alterations. The upregulation of disaccharides (trehalose, maltose, and sucrose) in grain tissue indicates enhanced carbon partitioning and storage [39,40]. Trehalose is known for its role in stress tolerance and cellular protection. It showed strong binding interactions with multiple upregulated proteins, including seed storage proteins and stress-responsive enzymes, suggesting a coordinated metabolic regulation. Upregulated amino acids, particularly alanine and aspartic acid, reflect enhanced nitrogen metabolism and protein synthesis. Jasmonic acid accumulation in both grain and root tissues indicates activation of defense signaling pathways and improved stress responsiveness [41,42]. The pathway analysis revealed significant modulation of aminoacyl-tRNA biosynthesis and carbon fixation pathways, which provide mechanistic insights into enhanced protein synthesis and photosynthetic efficiency that led to improved plant growth and yield.
The molecular docking analysis provided novel insights into functional protein–metabolite interactions, particularly between seed storage proteins and disaccharides. Multiple strong binding affinities observed suggest that metabolite accumulation may regulate protein function through direct binding interactions [43]. Strong binding for helicase-sugar complexes [ΔG −7.8 Kcal/mol for A0A3B6LIU4-Sucrose; Table S11 and Figure 4c] suggests that sucrose may directly fuel DNA repair mechanisms. Concurrent upregulation of helicases [Table 1 and S1-4] and sugars [Figure 1a and 2a] alongside predicted helicase-sugar interactions indicates coordinated energy utilization for genomic stability. While trehalose and other disaccharides accumulate during seed maturation and are known to stabilize proteins during desiccation through osmolyte-based mechanisms, the specific interaction between helicases and sucrose has not been characterized experimentally. Bifunctional inhibitor A0A3B6MXN3 binding to jasmonic acid (ΔG −7.2 Kcal/mol) links defense signaling directly to protease regulation [44]. Jasmonic acid preferentially binds to defense and LTP. Strong binding affinity of aspartic proteinase A0A3B6A2J7 toward maltose (ΔG −7.2 Kcal/mol) connects sugar metabolism and protein turnover. Similarly, the interaction of disaccharides with ATP synthase and plasma-membrane ATPase enzymes suggests a role of carbohydrate reserves in cellular energy production. Defense proteins such as defensins and Bowman-Birk inhibitors also showed strong trehalose binding, which might support their stabilization under desiccation. Further, peroxidases were predicted to interact with stearic acid and allantoin, suggesting a synergistic antioxidative defense. Together, molecular docking data emphasize a network-level crosstalk between sugar metabolism, membrane energization, and stress-hormone signaling during seed maturation in PGPB/AMF-treated wheat [45].
The comparison between 1st and 2nd-year field trials revealed consistent as well as variable protein expression patterns, reflecting the dynamic nature of plant–microbe interactions under various environmental conditions. Proteins such as bifunctional inhibitor/plant LTP/seed storage helical domain-containing proteins and serpins showed consistent upregulation in both seasons. Variable expression of some proteins (e.g., cold-regulated proteins and heat shock proteins) may reflect adaptive responses to year-specific environmental conditions, suggesting the flexibility of microbial-mediated stress tolerance mechanisms in plants. Metabolomics across both growing seasons showed reproducible and consistent metabolomic patterns for the common treatments (CP4, AMF, CP4+AMF). In both years, CP4+AMF consistently induced the most comprehensive upregulation in wheat grain and roots. This was characterized by coordinated elevation of sugars (sucrose, glucose, and trehalose), polyols (mannitol, arabinitol, and xylitol), and amino acids (alanine, serine, methionine, and valine) as evident in the univariate analyses and correlation matrices. VIP analyses from both years highlighted overlapping discriminant features, including trehalose, L-methionine, and glycolytic intermediate (pyruvate), alongside antioxidant (ascorbate). Single inoculations showed consistent but narrower responses: CP4 preferentially elevated glycolytic endpoints and short-chain acids while AMF increased selected sugars and aromatic pathway precursors. The tripartite treatment in year 2 revealed a novel synergistic signature that substantially amplified the established CP4+AMF metabolic module. It strengthened the positive correlation networks among central carbon and amino acid metabolites. The tripartite treatment elevated additional VIP contributors beyond the bipartite combination, indicating that AHP3 addition creates an additive enhancement rather than a distinct metabolic reprogramming. The metabolic pathway analysis identified common pathways modulated in wheat grains during both years. Glyoxylate and dicarboxylate metabolism, aminoacyltRNA biosynthesis, amino acid metabolism, and galactose metabolism were significantly modulated in response to PGPB/AMF treatment. Glyoxylate and dicarboxylate metabolism enrichment [impact 0.22, FDR <0.05; Table 2] is reflected in the upregulation of specific metabolites, including glyceric acid, oxalic acid, and pyruvic acid [Figure 2].
The proteomic analysis was limited to grain tissue; however, root proteomics could provide additional insights into microbial colonization and nutrient uptake mechanisms [46]. Although molecular docking analysis is informative, it requires further refined computational analyses to check the binding stability as well as experimental validation to confirm protein–metabolite interactions under physiological conditions [47]. In addition, this study focused on a single wheat cultivar (HD-3086) and specific environmental conditions, limiting the generalizability of findings [48]. Temporal dynamics of protein and metabolite changes during grain development, analysis of root proteomics, and validation of key protein–metabolite interactions through biochemical assays may be investigated in the future. Multi-cultivar studies across diverse environmental conditions would help establish the broader applicability of these microbial consortia [49]. Further, integration of transcriptomic data would provide a more complete understanding of gene regulation underlying the observed protein and metabolite changes. The molecular insights gained from this study support the practical application of microbial consortia in sustainable wheat production [50]. The identification of key proteins and metabolites involved in growth and yield enhancement provides molecular markers for monitoring treatment efficacy. The indication of enhanced stress tolerance mechanisms at the molecular level supports the use of these consortia in sustainable agricultural practices.
5. CONCLUSION
This study provides comprehensive molecular insights into the mechanisms underlying wheat improvement through PGPB and AMF consortia treatment observed under field conditions (Yadav et al., 2022a). The tripartite microbial combination (CP4+AHP3+AMF) showed superior performance, upregulating 88 proteins in grain tissue and significantly altering metabolite profiles in both grain and root tissues. Based on proteomic, metabolomic, and interactomic analyses, we suggest that wheat enhancement through PGPB/AMF treatment is driven by three interconnected biochemical systems. First, enhanced protein accumulation (prolamins, non-specific LTP, and enzymatic inhibitors) is supported by an upregulated aminoacyl-tRNA biosynthetic pathway and significant amino acid accumulation (≥2-fold increases in alanine, methionine, aspartate, and glutamate). This was complemented by EC protein I/II upregulation (2.7-fold in tripartite), indicating improved zinc ion homeostasis and micronutrient biofortification aligned with field observations. Second, altered energy metabolism is evident in significantly upregulated ATP synthase subunit O and plasma membrane ATPase (>9-fold in tripartite) indicates elevated energy production capacity. This was accompanied by coordinated metabolomic changes: Tripartite treatment induced ≥2-fold increases in energy storage sugars (trehalose, maltose, and sucrose) and polyols (mannitol, arabinitol, and xylitol). Pathway analysis confirmed significant modulation of glyoxylate and dicarboxylate metabolism which replenishes tricarboxylic acid cycle intermediates for sustained energy and biosynthetic requirements. Third, integrated stress tolerance through redox homeostasis in PGPB/AMF-treated wheat. This was evident in upregulation of antioxidant enzymes such as superoxide dismutase (4.2-fold in CP4- and 3.1-fold in tripartite) and peroxidase. sHSPs (both cytoplasmic and chloroplastic) were universally upregulated. They might function as molecular chaperones during biosynthetic stress. Metabolomic evidence included ≥2-fold increases in allantoin (a ROS scavenger) and jasmonic acid (a stress hormone), integrating defense signaling with redox management. Further, defense proteins (defensins, protease inhibitors, and WRKY factors) showed consistent upregulation, indicating comprehensive stress priming. These coordinated changes suggest that nutrient availability provides biosynthetic substrates, energy mobilization powers biosynthesis, and antioxidant/defense systems protect metabolic machinery during high metabolic activity. The highly upregulated histones and helicases across PGPB/AMF treatments indicate transcriptional reprogramming in wheat seeds at maturity. Strong binding affinities predicted for helicase-sucrose/maltose complexes suggest substrate channeling for enhanced energy requirements during nucleic acid processing and demand further validation. These findings establish a molecular framework for understanding microbial consortia-mediated wheat improvement and support their application in sustainable agriculture. The identification of key proteins, metabolites, and pathways in wheat HD3086 may serve as molecular markers for optimizing microbial treatments and monitoring their efficacy under field conditions.
6. AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
7. FUNDING
The authors acknowledge funding by the Science and Engineering Board (SERB), Department of Science and Technology, Government of India (grant number EMR/2016/006311), including Junior and Senior Research Fellowship to Pankaj Ror. Junior and Senior Research Fellowship to Radheshyam Yadav was awarded by the University Grants Commission (UGC), New Delhi, India.
8. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
9. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
10. DATA AVAILABILITY
The mass spectrometry proteomics data generated for this study have been deposited in the PRIDE Archive (https://www.ebi.ac.uk/pride/) under accession number PXD035742.
11. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
12. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declare that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
13. SUPPLEMENTARY MATERIAL
The supplementary material can be accessed at the link here: https://jabonline.in/admin/php/uploadss/1459_pdf.pdf
REFERENCES
1. Food and Agriculture Organization. Agricultural Production Statistics. 2000-2021. FAOSTAT Analytical Brief Series No. 60. Rome: FAO;2022. [CrossRef]
2. Ramakrishna W, Yadav R, Li K. Plant growth promoting bacteria in agriculture: Two sides of a coin. Appl Soil Ecol. 2019;138:10-8. [CrossRef]
3. Ramakrishna W, Rathore P, Kumari R, Yadav R. Brown gold of marginal soil: Plant growth promoting bacteria to overcome plant abiotic stress for agriculture, biofuels and carbon sequestration. Sci Total Environ. 2020;711:135062. [CrossRef]
4. Olanrewaju OS, Ayangbenro AS, Glick BR, Babalola OO. Plant health: Feedback effect of root exudates-rhizobiome interactions. Appl Microbiol Biotechnol. 2019;103:1155-66. [CrossRef]
5. Liu Q, Cheng L, Nian H, Jin J, Lian T. Linking plant functional genes to rhizosphere microbes: A review. Plant Biotechnol J. 2023;21:902-17. [CrossRef]
6. Calzavara AK, Paiva PH, Gabriel LC, Oliveira AL, Milani K, Oliveira HC, et al. Associative bacteria influence maize (Zea mays L.) growth, physiology and root anatomy under different nitrogen levels. Plant Biol (Stuttg). 2018;20:870-8. [CrossRef]
7. Vuolo F, Novello G, Bona E, Gorrasi S, Gamalero E. Impact of plant-beneficial bacterial inocula on the resident bacteriome: Current knowledge and future perspective. Microorganisms. 2022;10:2462. [CrossRef]
8. Yang M, Gao X, Dong J, Gandhi N, Cai H, Von Wettstein DH, et al. Pattern of protein expression in developing wheat grains identified through proteomic analysis. Front Plant Sci. 2017;8:962. [CrossRef]
9. Vannini C, Domingo G, Fiorilli V, Seco DG, Novero M, Marsoni M, et al. Proteomic analysis reveals how pairing of a Mycorrhizal fungus with plant growth-promoting bacteria modulates growth and defense in wheat. Plant Cell Environ. 2021;44:1946-60. [CrossRef]
10. Dhawi F, Datta R, Ramakrishna W. Mycorrhiza and PGPB modulate maize biomass, nutrient uptake and metabolic pathways in maize grown in mining-impacted soil. Plant Physiol Biochem. 2015;97:390-9. [CrossRef]
11. Singh M, Kumar A, Singh R, Pandey KD. Endophytic bacteria: A new source of bioactive compounds. 3 Biotech. 2017;7:315. [CrossRef]
12. Etesami H, Jeong BR, Glick BR. Contribution of arbuscular mycorrhizal fungi, phosphate-solubilizing bacteria, and silicon to P uptake by plant. Front Plant Sci. 2021;12:699618. [CrossRef]
13. McLaughlin S, Zhalnina K, Kosina S, Northen TR, Sasse J. The core metabolome and root exudation dynamics of three phylogenetically distinct plant species. Nat Commun. 2023;14:1649. [CrossRef]
14. Choi KR, Jang WD, Yang D, Cho JS, Park D, Lee SY. Systems metabolic engineering strategies: Integrating systems and synthetic biology with metabolic engineering. Trends Biotechnol. 2019;37:817-37. [CrossRef]
15. Dhawi F, Datta R, Ramakrishna W. Proteomics provides insights into biological pathways altered by plant growth promoting bacteria and arbuscular mycorrhiza in sorghum grown in marginal soil. Biochim Biophys Acta Proteins Proteomics. 2017;1865:243-51. [CrossRef]
16. De Souza RS, Armanhi JS, Arruda P. From microbiome to traits: Designing synthetic microbial communities for improved crop resiliency. Front Plant Sci. 2020;11:1179. [CrossRef]
17. Yadav R, Ror P, Rathore P, Ramakrishna W. Bacteria from native soil in combination with arbuscular mycorrhizal fungi augment wheat yield and biofortification. Plant Physiol Biochem. 2020;150:222-33. [CrossRef]
18. Yadav R, Ror P, Rathore P, Kumar S, Ramakrishna W. Bacillus subtilis CP4, isolated from native soil in combination with arbuscular mycorrhizal fungi promotes biofortification, yield and metabolite production in wheat under field conditions. J Appl Microbiol. 2021;131:339-59. [CrossRef]
19. Yadav R, Ror P, Beniwal R, Kumar S, Ramakrishna W. Bacillus sp. and arbuscular mycorrhizal fungi consortia enhance wheat nutrient and yield in the second-year field trial: Superior performance in comparison with chemical fertilizers. J Appl Microbiol. 2022a;132:2203-19. [CrossRef]
20. Yadav R, Chakraborty S, Ramakrishna W. Wheat grain proteomic and protein-metabolite interactions analyses provide insights into plant growth promoting bacteria-arbuscular mycorrhizal fungi-wheat interactions. Plant Cell Rep. 2022b;41:1417-37. [CrossRef]
21. Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat Protoc. 2006;1:387-96. [CrossRef]
22. Pang Z, Chong J, Zhou G, De Lima Morais DA, Chang L, Barrette M, et al. MetaboAnalyst 5.0:Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021;49:W388-96. [CrossRef]
23. UniProt Consortium. UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 2023;51:D523-31. [CrossRef]
24. Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2023 update. Nucleic Acids Res. 2023;51(D1):D1373-80. [CrossRef]
25. Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243-50. [CrossRef]
26. Varadi M, Bertoni D, Magana P, Paramval U, Pidruchna I, Radhakrishnan M, et al. Alphafold protein structure database in 2024:Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024;52(D1):D368-75. [CrossRef]
27. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583-9. [CrossRef]
28. Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455-61. [CrossRef]
29. Kaur S, Suseela V. Unraveling arbuscular mycorrhiza-induced changes in plant primary and secondary metabolome. Metabolites. 2020;10:335. [CrossRef]
30. Edqvist J, Blomqvist K, Nieuwland J, Salminen TA. Plant lipid transfer proteins: Are we finally closing in on the roles of these enigmatic proteins? J Lipid Res. 2018;59(8):1374-82. [CrossRef]
31. Yuan D, Wu X, Jiang X, Gong B, Gao H. Types of membrane transporters and the mechanisms of interaction between them and reactive oxygen species in plants. Antioxidants (Basel). 2024;13:221. [CrossRef]
32. Li L, Liu KH, Sheen J. Dynamic nutrient signaling networks in plants. Annu Rev Cell Dev Biol. 2021;37:341-67. [CrossRef]
33. Dong C, Huang TC, Roberts TH. Genes encoding structurally conserved serpins in the wheat genome: Identification and expression profiles during plant development and abiotic and biotic stress. Int J Mol Sci. 2023;24(3):2707. [CrossRef]
34. Ferreira MM, Santos AS, Santos AS, Zugaib M, Pirovani CP. Plant serpins: Potential inhibitors of serine and cysteine proteases with multiple functions. Plants (Basel). 2023;12:3619. [CrossRef]
35. Ostergaard H, Rasmussen SK, Roberts TH, Hejgaard J. Inhibitory serpins from wheat grain with reactive centers resembling glutamine-rich repeats of prolamin storage proteins. Cloning and characterization of five major molecular forms. J Biol Chem. 2000;275(43):33272-9. [CrossRef]
36. Hathurusinghe SH, Azizoglu U, Shin JH. Holistic approaches to plant stress alleviation:A comprehensive review of the role of organic compounds and beneficial bacteria in promoting growth and health. Plants (Basel). 2024;13:695. [CrossRef]
37. Zan T, Li L, Li J, Zhang L, Li X. Genome-wide identification and characterization of late embryogenesis abundant protein-encoding gene family in wheat:Evolution and expression profiles during development and stress. Gene. 2020;736:144422. [CrossRef]
38. Shao W, Chen W, Zhu X, Zhou X, Jin Y, Zhan C, et al. Genome-wide identification and characterization of wheat 14-3-3 genes unravels the role of TaGRF6-A in salt stress tolerance by binding MYB transcription factor. Int J Mol Sci. 2021;22(4):1904. [CrossRef]
39. Lunn JE, Delorge I, Figueroa CM, Van Dijck P, Stitt M. Trehalose metabolism in plants. Plant J. 2014;79:544-67. [CrossRef]
40. Liu Y, Wang X, Ouyang L, Yao R, Wang Z, Kang Y, et al. Genome-wide analysis of trehalose-6-phosphate phosphatase gene family and their expression profiles in response to abiotic stress in groundnut. Plants (Basel). 2024;13:1056. [CrossRef]
41. Takagi H, Ishiga Y, Watanabe S, Konishi T, Egusa M, Akiyoshi N, et al. Allantoin, a stress-related purine metabolite, can activate jasmonate signaling in a MYC2-regulated and abscisic acid-dependent manner. J Exp Bot. 2016;67:2519-32. [CrossRef]
42. Kaur H, Chowrasia S, Gaur VS, Mondal TK. Allantoin: Emerging role in plant abiotic stress tolerance. Plant Mol Biol Rep. 2021;39:648-61. [CrossRef]
43. Kovalchuk N, Smith J, Bazanova N, Pyvovarenko T, Singh R, Shirley N, et al. Characterization of the wheat gene encoding a grain-specific lipid transfer protein TdPR61, and promoter activity in wheat, barley and rice. J Exp Bot. 2012;63:2025-40. [CrossRef]
44. Figueiredo L, Santos RB, Figueiredo A. Defense and offense strategies: The role of aspartic proteases in plant-pathogen interactions. Biology (Basel). 2021;10(2):75. [CrossRef]
45. Zhuang Y, Wang H, Tan F, Wu B, Liu L, Qin H, et al. Rhizosphere metabolic cross-talk from plant-soil-microbe tapping into agricultural sustainability: Current advance and perspectives. Plant Physiol Biochem. 2024;210:108169. [CrossRef]
46. Khoso M, Wagan S, Alam I, Hussain A, Ali Q, Saha S, et al. Impact of plant growth-promoting rhizobacteria (PGPR) on plant nutrition and root characteristics: Current perspective. Plant Stress. 2024;11:100341. [CrossRef]
47. Uroz S, Courty PE, Oger P. Plant symbionts are engineers of the plant-associated microbiome. Trends Plant Sci. 2019;24:905-16. [CrossRef]
48. Chialva M, Lanfranco L, Bonfante P. The plant microbiota: Composition, functions, and engineering. Curr Opin Biotechnol. 2022;73:135-42. [CrossRef]
49. Riggs PJ, Chelius MK, Iniguez AL, Kaeppler SM, Triplett EW. Enhanced maize productivity by inoculation with diazotrophic bacteria. Funct Plant Biol. 2001;28:829-36. [CrossRef]
50. Qiu Q, Bender SF, Mgelwa AS, Hu Y. Arbuscular mycorrhizal fungi mitigate soil nitrogen and phosphorus losses:A meta-analysis. Sci Total Environ. 2021;807:150857. [CrossRef]