1. INTRODUCTION
The use of Terminalia bellirica dates back thousands of years, and this herb has been in high demand among traditional medicine practitioners worldwide [1]. T. bellirica (Beleric) is a tropical fruit with a high level of bioactive compounds of pharmaceutical value [2]. Its antibacterial, antioxidant, and anticancer effects have been established recently, which has further established its universal applicability as a natural pharmacological source. Negligible literature has been reported on the cytotoxic or anticancer effects of the hydroalcoholic seed extracts of the plant, especially against breast cancer cell lines, although its antioxidant, antimicrobial, and anti-inflammatory properties had been extensively investigated [3]. Here, liquid chromatography-mass spectrometry (LC-MS)-based phytochemical profiling and Soxhlet-induced seed extraction techniques were used in the analysis of bioactive constituents [4], which were later predicted and evaluated with reference to drug-likeness in order to gain insight into the therapeutic value of the bioactive compounds.
T. bellirica harbors a number of effective antioxidant molecules, including polyphenols, tannins (gallic acid, ellagic acid, and chebulic acid), flavonoids, and gallotannins that exhibited high free radical scavenging properties in both DPPH and FRAP assays [5]. These compounds induced cellular antioxidant pathways (e.g., Nrf2, MAPK/NF-kB, and Akt/AMPK) and played a role in hepatoprotective, anti-inflammatory, and antidiabetic functions. The antimicrobial activity also had a broad spectrum of the plant extracts, with the methanolic and aqueous fruit extracts exhibiting significant inhibition zones and low MIC values against pathogenic bacteria, including drug-resistant strains such as MRSA, ESBL-producing Escherichia coli, and multidrug-resistant Pseudomonas aeruginosa [6]. Moreover, the T. bellirica extracts showed selective cytotoxicity to liver (HepG2) and lung (A549) carcinoma cell lines; tumor growth was inhibited by tannin-containing fractions, which regulated ERBB, PI3K-Akt, and MAPK pathways [7]. Cisplatin and doxorubicin (synergetic effect) were also reported with CI < 1 and low toxicity to normal cell lines [8].
Despite the documented cytotoxicity of T. bellirica fruit extracts, the anticancer activity of its hydroalcoholic seed extracts against the aggressive triple-negative MDA-MB-231 breast cancer cell line has not been well studied. Moreover, a comprehensive integration of in silico approaches, such as network pharmacology and molecular dynamics (MD), to identify the bioactive compounds and their mechanisms of action has been lacking. Therefore, this study proposed that the hydroalcoholic seed extract of T. bellirica had an anti-breast cancer effect on MDA-MB-231 cells through specific phytocompounds that target oncogenic signaling pathways. This was tested through LC-MS profiling, computational modeling, and in vitro validation using MTT assay. Considering the difficulty of phytochemical interactions in cancer biology, an in silico model provided a rational method to predict and analyze molecular interactions between bioactive compounds and cancer-associated targets. This aided in identifying the most promising candidates for further validation through in vitro studies. An in silico approach incorporated the evaluation of ADME properties as well as drug-likeness and toxicological profiles of the identified phytocompounds [9,10]. This was followed by molecular docking studies to examine the binding affinities of compounds against different target proteins involved in breast cancer. Further evaluation of ligand-receptor interactions was performed using MD simulations [11]. Herein, a fastidious experimental and computational pipeline was presented to screen conceivable bioactive candidates from T. bellirica for possible anti-breast cancer therapeutics. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) bioassay, which assesses cell viability, confirmed this bioactivity. The present study aimed to evaluate the anticancer potential of hydroalcoholic extracts of T. bellirica seeds against breast cancer cells and to elucidate their molecular mechanisms through an integrated in silico approach. It was hypothesized that the T. bellirica seed extract showed anticancer activity against breast cancer through in silico studies by interacting with the key target protein, and this was further proved by MTT assay. The objectives of this work were to prepare and characterize the hydroalcoholic extract of T. bellirica seeds, to identify and evaluate active compounds through in silico approaches targeting key breast cancer proteins, and finally to assess its activity against breast cancer cell lines.
2. MATERIALS AND METHODOLOGY
2.1. Seed Collection and Extraction
Seeds of T. bellirica, a very important medicinal tree of the Combretaceae family, were collected from Yuvika Herbs. All these seeds were thoroughly inspected for their morphological characteristics, such as shape, size, and texture, as per standard identification protocols of botany [12]. The study seed was identified and authenticated by Dr. K. Madhava Chetty, from the Department of Botany, Sri Venkateswara University, Tirupati, India. The collected seeds were thoroughly cleaned, dried, and stored in controlled conditions to maintain seed viability and homogeneity for future studies [13].
Seeds of T. bellirica were obtained, authenticated, and processed for extraction. A modified Soxhlet extraction method employing organic solvents was considered best for maximum phytocompound extraction [14,15]. The extraction of T. bellirica seeds was carried out with 100% ethanol. Three hundred milliliters of the solvent were used to extract 15 g of strictly suspended T. bellirica seed powder. A Soxhlet apparatus was used for extraction for up to 3 h, until the solvent in the extractor siphon tube becomes colorless. The temperature was initially maintained at 30°C as the solvent started to boil, with a gradual increase in temperature to 70°C without exceeding the boiling point. The extract was concentrated at 45°C while a rotary evaporator was used for 45 min and lyophilized, and the dry crude concentrate was stored at −4°C for in vitro anticancer activity and LC-MS data analysis [16].
2.2. LC-MS Analysis
LC-MS analysis of T. bellirica was conducted on a Waters 2645 separation module with a Waters Micromass ZQ mass spectrometer. The mobile phases consisted of 0.1% formic acid in water (Mobile Phase A) and 0.1% formic acid in acetonitrile (Mobile Phase B). Sample preparation consisted of the solubilization of 20 mg of the dry powder in methanol, followed by adjusting the final volume to 25 mL, followed by filtration with a 0.22 μm syringe filter to remove particulates. Analysis had a 10 μL injection volume. The gradient program proceeded as follows: The initial flow of 0.500 mL/min with 90% Mobile Phase-A, along with 10% Mobile Phase B, was switched to 5% Mobile Phase A and 95% Mobile Phase-B at 7.00 min, remaining the same until 12.00 min. The flow switched back to the initial conditions by 13.00 min and remained the same until the end of the run at 15.00 min. The column was an Accucore C18 (50 × 4.6 mm, particle size of 5 μm) from the company Thermo Scientific, maintained at 40°C. The mass spectrometer operated under positive electrospray ionization (ES+) conditions with the capillary voltage set as 2.8 kV, the cone voltage as 30 V, the source temperature as 140°C, as well as the desolvation temperature as 400°C, with the desolvation gas set at 600 L/h. Full scan data were acquired over the mass range of m/z 100–1000. Data processing occurred with the use of the MassLynx V4.1 software that can permit detailed examination of the resultant mass spectra.
The work starts with the selection of phytocompounds to be used as inferences from an LC-MS analysis of the samples. All of these compounds were obtained from a formatted library of plant-derived metabolites. The molecular specifications of the compounds, for example, using ChemDraw and MolSoft (MolSoft Drug Likeness and Molecular Properties Software), were determined through computational tools and software incorporated into the study. The molecular formula, molecular weight, HBA, HBD, MolLogP, MolLogS, molecular polar surface area (MolPSA), and molecular volume (MolVol) were calculated through standard algorithms integrated into these software platforms. The pKa values of the basic and acidic groups were predicted through MarvinSketch, and blood–brain barrier (BBB) penetration scores were derived through the brain or intestinal estimated permeation (BOILED-Egg) model.
The MolSoft drug-likeness scoring function judges each of these compounds with respect to the drug-likeness properties because the function describes the compatibility of molecular properties with respect to standard pharmacokinetic and pharmacodynamic profiles. Positive scores indicate an increased chance for that compound to be a drug candidate [17,18].
To strengthen its accuracy and replicability, measurements were made in triplicate for every single observation, and the results were considered using simple statistical methods. Positive drug-likeness scores were used to study other pharmacological aspects of these compounds in terms of the overall balance between hydrophilicity, lipophilicity, and other parameters that seem very important.
2.3. Toxicological Properties of the Phytocompounds
The ProTox-II database was used for the online in silico prediction of the toxicological effects of 13 phytocompounds. An assessment of the toxicological effects of the phytocompounds, such as their carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity, was performed [19]. The ProTox-II database utilizes machine learning models to predict toxicity profiles, providing insight into possible undesirable effects. The results for each compound were recorded systematically, and the data were further analyzed to identify any patterns or correlations between the phytocompounds and their toxicological profiles.
2.4. ADME Properties of the Phytocompounds
Each compound’s ADME properties were calculated through the Swiss ADME database. The ADME properties of the phytochemicals were evaluated to assess their drug-likeness and pharmacokinetic potential. Parameters such as gastrointestinal absorption, BBB permeability, P-glycoprotein substrate, and the major cytochrome P450 inhibitors CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 were assessed [20]. Drug likeness was derived from Lipinski’s rule of five, with the number of rule violations noted for each. The analysis was carried out using validated in silico tools and databases. GI absorption and BBB permeability were used to assess bioavailability and central nervous system penetration, whereas Pgp substrate and CYP enzyme inhibition data would have added to the knowledge of possible interactions and metabolic stability. The data were organized systematically to indicate the extent of adherence of each compound to Lipinski’s rule.
2.5. Target Identification
Canonical SMILESs were analyzed for target prediction through Swiss Target Prediction (http://swisstargetprediction.ch/), aligning them with known therapeutic drug molecules [21]. To ensure the robustness of the network and avoid spurious links, clear thresholds were applied for target inclusion. For the phytocompounds, targets were predicted through Swiss Target Prediction, and only those with a probability score >0.1 were retained. In addition, target proteins associated with breast cancer were identified on the basis of documented targets sourced from the GeneCards Database (https://www.genecards.org/). For breast cancer-associated targets from the GeneCards database, a relevance score threshold of >10 was applied, focusing the analysis on the top 1842 high-confidence genes. To identify combined genes, compound-related genes and disease-related genes were merged, and the overlap was visualized through a Venn diagram.
2.6. Pathway and Network Analysis
The STRING database (https://string-db.org/) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (https://www.genome.jp/kegg/) were used to investigate protein–protein interactions (PPIs) and molecular pathways associated with protein targets related to BC [22]. The relationships among compounds, target proteins, and pathways relevant to breast cancer were visualized through Cytoscape v3.6.1 (https://cytoscape.org/) [23]. Network visualization was enhanced by employing a color scale and varying node sizes, where the size of each node corresponded to its edge count (number of connections). Nodes with higher edge counts were represented as larger nodes, highlighting their significance in the network.
2.7. Molecular Docking
The present investigation employed the molecular docking of ligands against the target protein through AutoDock Vina. First, Open Babel software was employed to generate the three-dimensional structures of the ligands for docking after they were obtained through PubChem. Preparations for docking were also conducted for the receptor, the target protein. The grid box for docking was centered on the centroid of the co-crystallized ligand (coordinates: x = 26.5, y = 20.8, z = 49.1) with dimensions set to 60 × 60 × 60 Å. The exhaustiveness was set to 32. All other parameters were kept at their default values. Furthermore, duplicate docking procedures were initiated. The binding affinities of the resulting docking locations were assessed, and the optimal poses were selected for further analysis. The PyMOL algorithm was utilized to analyze the ligand-receptor interactions and binding mechanism. The efficacy of the medicines and phytochemicals as inhibitors of the target protein was evaluated by docking analysis [24].
2.8. MD
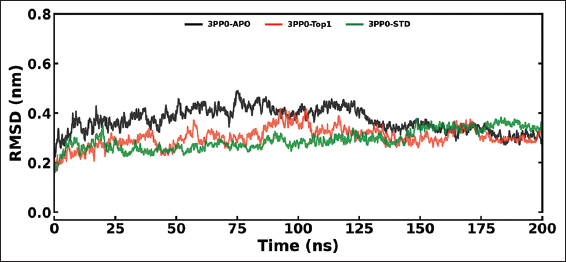
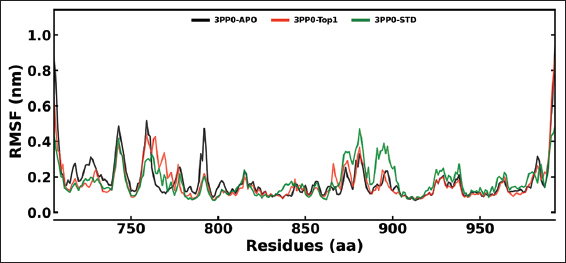
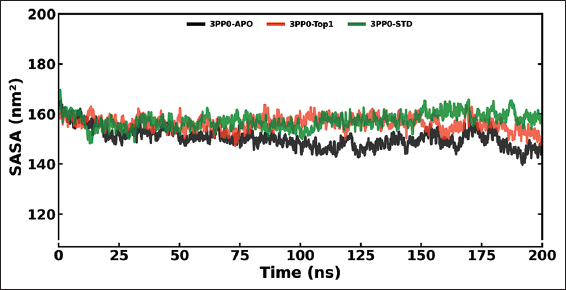
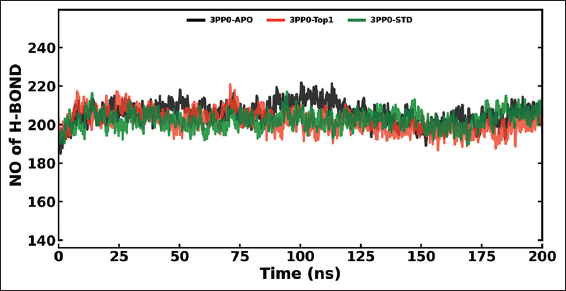
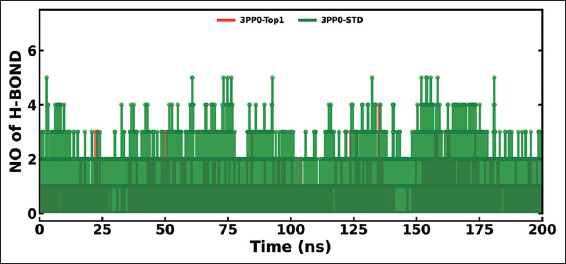
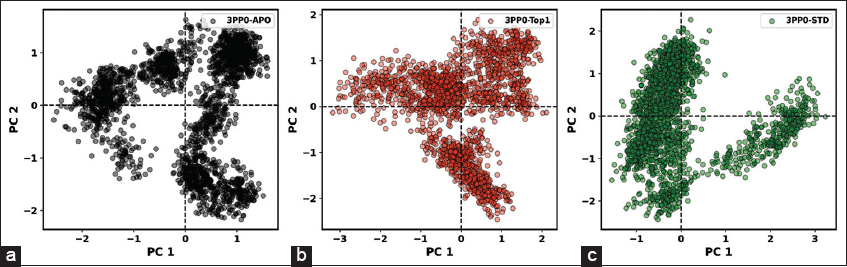
The MD simulations were conducted on the kinase domain structure of human HER2, the ErbB2 protein (PDB ID: 3PP0), in complex with ligands of first interest obtained from the docking investigations. The ligand topologies were prepared with the ATB server. The GROMACS pdb2gmx module added hydrogen atoms to the molecules, as well as generated the protein topologies with the CHARMM36 force field. The systems prepared were initially energy-minimized in a vacuum with the steepest descent algorithm over 50,000 steps to remove steric clashes. The systems were then solvated in a cubic periodic box with at least 1.0 nm distance between the protein and the box edge using the simple point charge water model. The proper quantities of Na+ and Cl- counterions were introduced to neutralize the system as well as to give a physiological salt concentration of 0.15 M [25]. The systems were equilibrated over two stages: Initially, under an NVT (constant number of particles, volume, and temperature) ensemble for 100 ps at 300 K with the v-rescale thermostat, followed by one NPT (constant number of particles, pressure, and temperature) ensemble for 100 ps at 1 bar with the Parrinello–Rahman barostat. The final 200 ns MD production run was then simulated under the NPT ensemble. The long-range electrostatics were calculated with the Particle Mesh Ewald method with a cutoff of 1.0 nm on the short-range non-bonded interaction. The bond lengths were constrained with the LINCS algorithm. Trajectory analysis, root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and hydrogen bonding (H-bonds) were carried out with the help of GROMACS analysis tools. The binding free energy has also been calculated with the help of the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method [26]. For improved accuracy for 3PP0-STD and 3PP0-TOP1, the final calculations were carried out using the last 50 ns (for every 1000 frames) [27].
2.9. MTT Assay
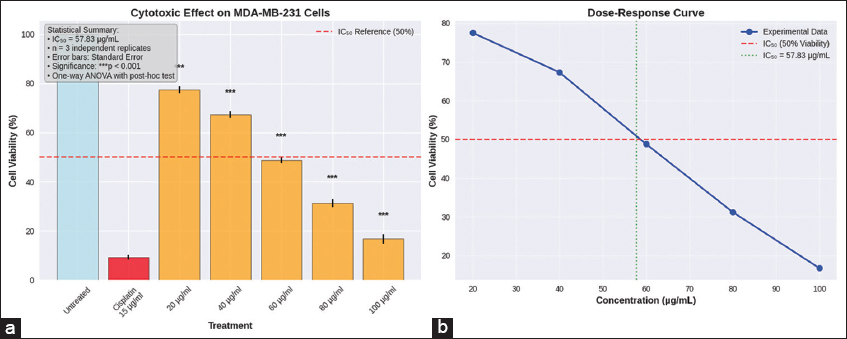
The T. bellirica extract-induced cytotoxic effect on MDA-MB-231 cells was assessed with the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The hydroalcoholic extract dissolved in DMSO was used, with the final DMSO concentration in all the treatment wells, including the vehicle control, never to exceed 0.1% (v/v). The cells were seeded in 96-well plates and, after 24 h, treated with a range of the extract concentration (20–100 μg/mL), vehicle control (0.1% DMSO), or positive control (10 μM cisplatin) for 24 h. Following the treatment, the medium was removed and replaced with fresh medium with MTT at the final working concentration of 0.5 mg/mL. The plates were incubated for 4 h at 37°C to permit the formation of the formazan crystals. The formazan crystals were then dissolved in the wells with the help of 100 μL DMSO per well. The absorbance measurement occurred at 570 nm with a reference at 690 nm in a microplate spectrophotometer. The experiment was undertaken in three independent biological replicates, each one with six technical replicates (n = 3) [24].
2.10. Statistical Analysis
Each experiment was done in three independent biological repetitions. The data were given in mean and standard deviation (SD). Values of cell viability produced during the MTT assay were transformed to the untreated control and indicated as percentage viability. The non-linear regression that employed a four-parameter logistic (4PL) model was used to estimate dose-response data to determine half-maximal inhibitory concentration (IC50) and the 95% confidence interval (CI). The one-way analysis of variance (ANOVA) was done to make statistical comparisons between groups, and then the Dunnett multiple comparisons post hoc test was done to evaluate the differences between the treated groups and the untreated control. P < 0.05 was regarded as statistically significant. All graphical analyses were done in the GraphPad Prism software [28].
3. RESULTS
3.1. Extraction Process Results
Following Soxhlet extraction, the obtained extract was concentrated using a rotary evaporator such that, upon complete evaporation, 5 g of solvent-free concentrated extract was obtained. Thereafter, the concentrated extract was subjected to LC-MS analyses. Ethanol was selected as the extraction solvent because it efficiently extracted T. bellirica constituents and effectively solubilized polar compounds.
3.2. LC-MS
LC–MS analysis was employed to identify phytoconstituents of T. bellirica seed extract. The matching was done using retention times, experimental m/z values, MS/MS fragmentation patterns, database/library matching, metabolite class, and proposed chemical structures. The mass spectra were obtained in the positive ionization mode, and the m/z values obtained were mostly between 60 and 622. Figure 1 shows the T. bellirica chromatogram. Table 1 enumerated the phytochemical constituents that were detected using HR-LC/MS. The 13 selected compounds were further analyzed because they were the highest chromatographic peaks and they were the big bioactive chemical groups, such as flavonoids and phenolic acids. The identification of key compounds, such as glabranin, tricin, and epigallocatechin, with distinct molecular weights and structural characteristics, demonstrated the enrichment of bioactive phytocompounds in the extract. Table 1 summarizes the identity confidence. In the absence of authentic reference standards, all identifications of compounds were tentatively identified (level of confidence 3) by accurate mass determination (mass error <5 ppm) and spectral similarity with in-house and external databases (GNPS and MassBank).
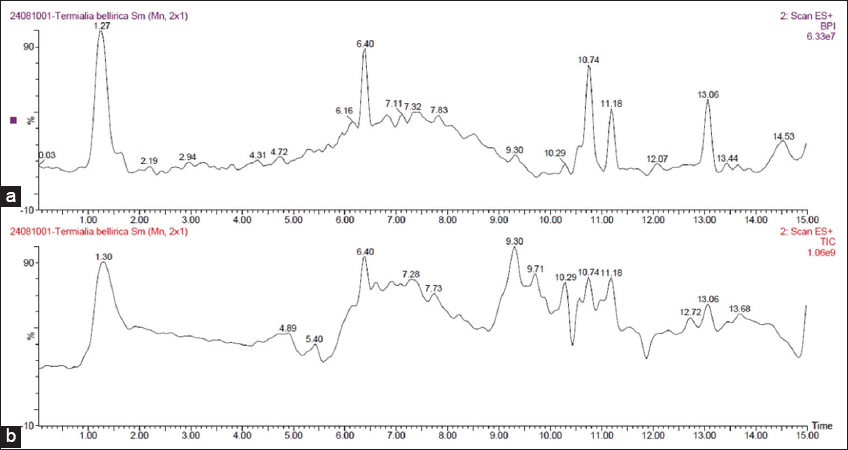 | Figure 1: Chromatogram of the identified phytochemical constituents of the hydroalcoholic extract of Terminalia bellirica through HR-LC-MS.
[Click here to view] |
Table 1: LC-MS analysis of the hydroalcoholic extract of Terminalia bellirica.
| Compound name | RT | Structure | Molecular formula | Molecular weight (g/mol) | Adduct | Compound class |
|---|
| 3’,5,7-Trihydroxy-4’- methoxy flavanone | 1.282 | 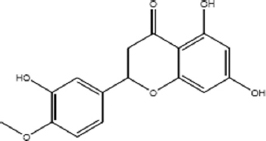 | C16H14O6 | 302.2788 | M-H | Flavonoid |
| Isoamylamine | 1.676 | 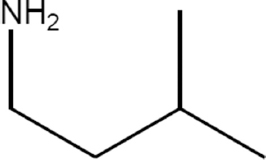 | C5H13N | 87.1634 | M+H | N/A |
| 4-Coumaroylquinic acid | 1.761 | 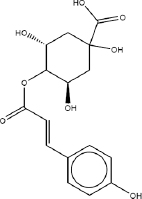 | C16H14O6 | 302.2788 | M-H | N/A |
| Glabranin | 6.361 | 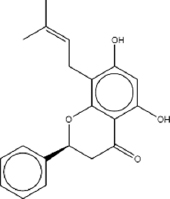 | C20H20O4 | 324.3704 | M+H | Flavonoids |
| Tricin | 6.601 |  | C17H14O7 | 330.2889 | M+H | Flavonoids |
| Quinate | 6.652 | 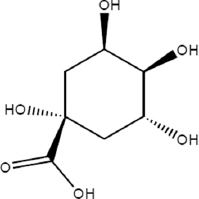 | C7H12O6 | 192.1666 | M-H | N/A |
| Phloretin-2’- O-glucoside | 7.114 |  | C21H24O10 | 436.4093 | M+H | N/A |
| 6-Prenylnaringenin | 7.712 |  | C20H20O5 | 340.3698 | M+H | N/A |
| Flavone | 7.729 |  | C15H10O2 | 592.5453 | M+H | Flavone C-glycosides |
| Epigallocatechin | 7.729 |  | C15H14O7 | 306.2675 | M+H | N/A |
| 1,4-bis (p-tolylamino) anthraquinone | 9.302 |  | C28H22N2O2 | 418.4865 | M+H | N/A |
| Eicosenoic acid | 10.65 |  | C18H34O2 | 310.5 | M+H | N/A |
| Fortunellin | 10.739 |  | C28H32O14 | 592.5453 | M+H | Flavonoid |
3.3. Molecular Properties and Drug-Likeness of the Phytocompounds
The pharmacological research goals were to examine the molecular properties and drug-likeness of the thirteen phytocompounds that were chosen to be included in the pharmacological research. Parameters such as molecular formula, molecular weight, HBAs, HBDs, MolLogP, MolLogS, MolPSA, MolVol, pKa, score on BBB, and number of stereocenters are calculated and presented in Table 2.
Table 2: Molecular properties and drug-likeness of the phytocompounds.
| S. NO | Name of the compound | Molecular formula | Molecular weight | Number of HBA | Number of HBD | MolLogP | MolLogS | Molecular polar surface area | Molecular volume | pKa of most basic/acidic group | Blood-brain barrier score | Number of stereo centers | Drug- likeness model score |
|---|
| 1 | Isoamylamine | C5 H13N | 87.10 | 1 | 2 | 1.03 | −0.23 (in Log (moles/L)) 51512.44 (in mg/L) | 21.67 A2 | 103.75 A3 | 10.30/22.57 | 3.87 | 0 | −1.37 |
| 2 | Glabranin | C20H2OO4 | 324.14 | 4 | 2 | 4.61 | −4.26 (in Log (moles/L)) 17.82 (in mg/L) | 52.98 A2 | 344.74 A3 | <0./8.82 | 3.99 | 1 | 0.91 |
| 3 | Tricin | C17H14O7 | 330.07 | 7 | 3 | 2.53 | −2.68 (in Log (moles/L)) 694.31 (in mg/L) | 86.69 A2 | 323.54 A3 | <0./6.70 | 2.74 | 0 | −0.08 |
| 4 | Phloretin-2’- O- glucoside | C21H24O10 | 436.14 | 10 | 7 (>5) | 0.45 | −1.87 (in Log (moles/L)) 5837.71 (in mg/L) | 144.94 A2 | 389.19 A3 | <0./8.23 | 1.70 | 5 | 0.66 |
| 5 | Flavone | C18H23NO3 | 301.17 | 4 | 4 | 2.42 | −1.61 (in Log (moles/L)) 7414.61 (in mg/L) | 62.62 A2 | 294.71 A3 | 9.31/10.09 | 3.60 | 2 | 1.21 |
| 6 | Epigallocatechin | C15H14O7 | 306.07 | 7 | 6 (>5) | 0.26 | −0.91 (in Log (moles/L)) 37829.65 (in mg/L) | 105.93 A2 | 271.81 A3 | <0./9.36 | 2.50 | 2 | −0.04 |
| 7 | 1,4-bis (p-tolylamino) anthraquinone | C28H22N2O2 | 418.17 | 2 | 2 | 8.15 (>5) | −6.06 (in Log (moles/L)) 0.36 (in mg/L) | 42.61 A2 | 414.06 A3 | −2.55/21.57 | 3.55 | 0 | −0.80 |
| 8 | Fortunellin | C28H32O14 | 592.18 (>500) | 14 (>10) | 7 (>5) | 0.82 | −1.79 (in Log (moles/L)) 9499.99 (in mg/L) | 172.17 A2 | 532.32 A3 | <0./9.77 | 1.24 | 10 | 0.70 |
| 9 | 3’,5,7-Trihydroxy- 4’- methoxy flavanone | C16H14O6 | 302.08 | 6 | | 2.51 | −2.77 (in Log (moles/L)) 511.01 (in mg/L) | 78.55 A2 | 283.84 A3 | <0./8.29 | 2.89 | 1 | 0.59 |
| 10 | 4-Coumaroylquinic acid | C16H18O8 | 338.10 | 8 | 5 | −0.02 | −1.34 (in Log (moles/L)) 15281.13 (in mg/L) | 111.98 A2 | 326.25 A3 | <0./4.69 | 2.20 | 2 | 0.03 |
| 11 | Quinate | C7H12O6 | 192.06 | 6 | 5 | −2.17 | −0.05 (in Log (moles/L)) 171548.56 (in mg/L) | 90.69 A2 | 172.85 A3 | <0./4.69 | 1.91 | 2 | −1.06 |
| 12 | 6-Prenylnaringenin | C20H2OO5 | 340.13 | 5 | 3 | 4.49 | −4.44 (in Log (moles/L)) 12.24 (in mg/L) | 69.85 A2 | 355.23 A3 | <0./8.82 | 2.95 | 1 | 1.36 |
| 13 | Eicosenoic acid | C20H38O2 | 310.29 | 2 | 1 | 8.12 (>5) | −5.79 (in Log (moles/L)) 0.51 (in mg/L) | 28.89 A2 | 384.40 A3 | <0./4.26 | 4.40 | 0 | −0.30 |
The MolSoft software was used in the generation of drug-likeness model scores of the reviewed compounds. Seven compounds were found to have positive drug-likeness scores and were chosen to undergo further pharmacological studies. The other six compounds (isoamylamine, tricin, epigallocatechin, 1,4-bis(p-tolylamino)anthraquinone, quinate, and eicosenoic acid) were eliminated because of poor drug-likeness scores, indicating poor oral bioavailability. This screening approach was based on the selection of compounds that had good physicochemical and pharmacokinetic attributes.
The evaluation of toxicological properties via the ProTox-II database and ADME characteristics through in silico tools yielded significant insights into the seven selected phytocompounds.
3.4. Toxicological Properties (ProTox-II Analysis)
The ProTox-II platform was used to determine the toxicology of the phytocompounds. The parameters predicted included carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity [Table 3]. Each of the seven compounds chosen had good toxicity profiles, and there was no significant evidence of carcinogenicity. Nobody anticipated significant immunotoxic or mutagenic potentials in a couple of compounds, and cytotoxicity levels were within acceptable ranges, which justifies their use in the subsequent research.
Table 3: Toxicological properties of the phytocompounds.
| S. No | Name of compound | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity |
|---|
| 1 | Isoamylamine | No | No | No | No |
| 2 | Glabranin | No | Yes | No | No |
| 3 | Tricin | No | No | No | No |
| 4 | Phloretin-2’- O-glucoside | No | No | No | No |
| 5 | Flavone | YES | No | No | Yes |
| 6 | Epigallocatechin | No | No | No | No |
| 7 | 1,4-bis (p-tolylamino) anthraquinone | Yes | No | Yes | No |
| 8 | Fortunellin | No | Yes | No | No |
| 9 | 3’,5,7-Trihydroxy-4’- methoxy flavanone | No | Yes | No | No |
| 10 | 4-Coumaroylquinic acid | No | Yes | No | No |
| 11 | Quinate | No | No | No | No |
| 12 | 6-Prenylnaringenin | No | No | No | No |
| 13 | Eicosenoic acid | No | No | No | No |
3.5. ADME Property Analysis
The ADME properties were assessed, and it was found that the majority of the seven compounds selected had high gastrointestinal absorption and moderate/good BBB permeability. The vast majority of compounds were not associated with P-glycoprotein substrates, which means that there is a lower probability of having bioavailability limitations in the form of efflux. There was limited cytochrome P450 inhibition [Table 4], which indicates a low likelihood of drug–drug interaction. Six of them fell under the Lipinski rule of five, and Fortunellin was disqualified because of several infractions.
Table 4: ADME properties of the phytocompounds.
| S. No | Name of compound | GI absorption | Blood–brain barrier permeant | Pgp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | LipinskiRule | Bioavailability score |
|---|
| 1 | Isoamylamine | High | Yes | No | No | No | No | No | No | Yes | 0.55 |
| 2 | Glabranin | High | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 3 | Tricin | High | No | No | Yes | No | Yes | Yes | Yes | Yes | 0.55 |
| 4 | Phloretin-2’- O- glucoside | Low | No | Yes | No | No | No | No | No | Yes | 0.55 |
| 5 | Flavone | High | Yes | No | Yes | Yes | No | No | No | Yes | 0.55 |
| 6 | Epigallocatechin | High | No | No | No | No | No | No | No | Yes | 0.55 |
| 7 | 1,4-bis (p-tolylamino) anthraquinone | High | No | No | Yes | Yes | No | No | No | Yes | 0.55 |
| 8 | Fortunellin | Low | No | Yes | No | No | No | No | Yes | No | 0.17 |
| 9 | 3’,5,7-Trihydroxy- 4’- methoxy flavanone | High | No | Yes | Yes | No | No | No | Yes | Yes | 0.55 |
| 10 | 4- Coumaroylquinic acid | Low | No | No | No | No | No | No | No | Yes | 0.56 |
| 11 | Quinate | Low | No | Yes | No | No | No | No | No | Yes | 0.56 |
| 12 | 6-Prenylnaringenin | High | No | No | Yes | No | Yes | Yes | Yes | Yes | 0.55 |
| 13 | Eicosenoic acid | Low | No | No | Yes | No | Yes | No | No | Yes | 0.85 |
To integrate findings from multiple databases, six phytocompound targets were retrieved from the Swiss Target Prediction database, whereas disease-associated targets were curated through the GeneCards database. The phytocompound targets were uploaded into the STRING database to study the PPI networks shown in Figure 2 and perform KEGG pathway analysis. PPI analysis revealed significant interaction clusters, shedding light on key biological processes and molecular pathways influenced by the selected phytocompounds. KEGG pathway analysis revealed pathways potentially modulated by these targets, many of which were associated with specific disease states.
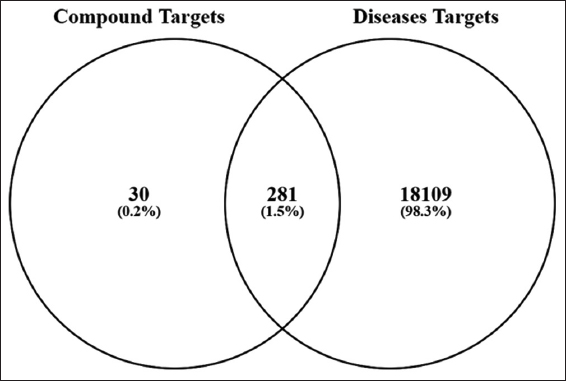
The Venn diagram juxtaposes the two datasets, “Compound Targets” (311 elements) and “Disease Targets” (18,390 elements). The analysis revealed that 281 targets (1.5%) were common to both lists, indicating potential shared biological mechanisms or interactions that could be critical for understanding therapeutic effects or drug development. The analysis also revealed that 30 targets (0.2%) are unique to the “Compound Targets” list, suggesting pathways or mechanisms that specifically appraise the compounds studied. Finally, 18,109 targets (98.3%) found only in the “Disease Targets” list indicate disease-related mechanisms not directly associated with the compounds. The large number of unique disease targets (18,109) reflects the complexity of breast cancer. However, by applying a high-confidence threshold (relevance score >10) to the GeneCards data, the analysis focused on 1,842 top-ranked targets for a more biologically relevant network. The 281 overlapping targets between this high-confidence set and the compound targets were considered the most promising for mediating the anti-breast cancer effects and were used for subsequent pathway analysis [Figure 3].
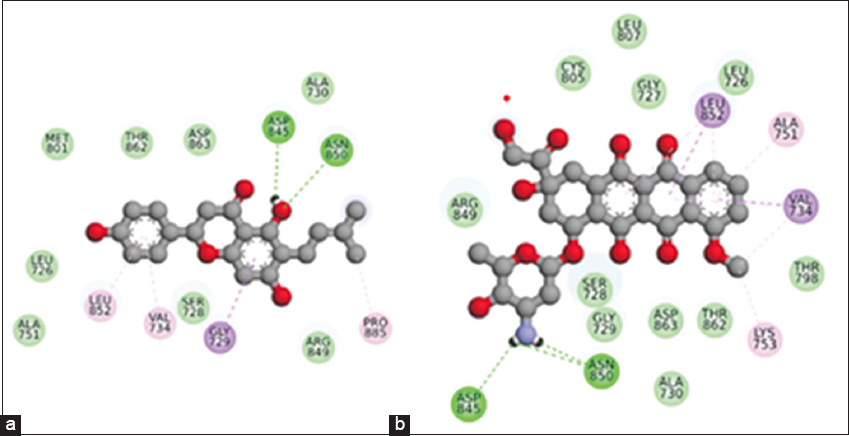
A complete network was built to describe the interactions between compound-specific targets and disease-associated targets, as shown in Figure 4 and Table 5. The network shows some key nodes and hubs to better appreciate the critical modulators in the system. The ligand-target interaction network is shown in yellow. The identified targets and their ligands were studied through molecular docking, which revealed compelling and specific interactions with important protein targets. To validate these interactions further, we performed MD simulations that revealed the stability and dynamic behavior of the docked complexes over time. These analyses confirmed worthwhile interactions and reinforced the therapeutic potential of the set of phytocompounds.
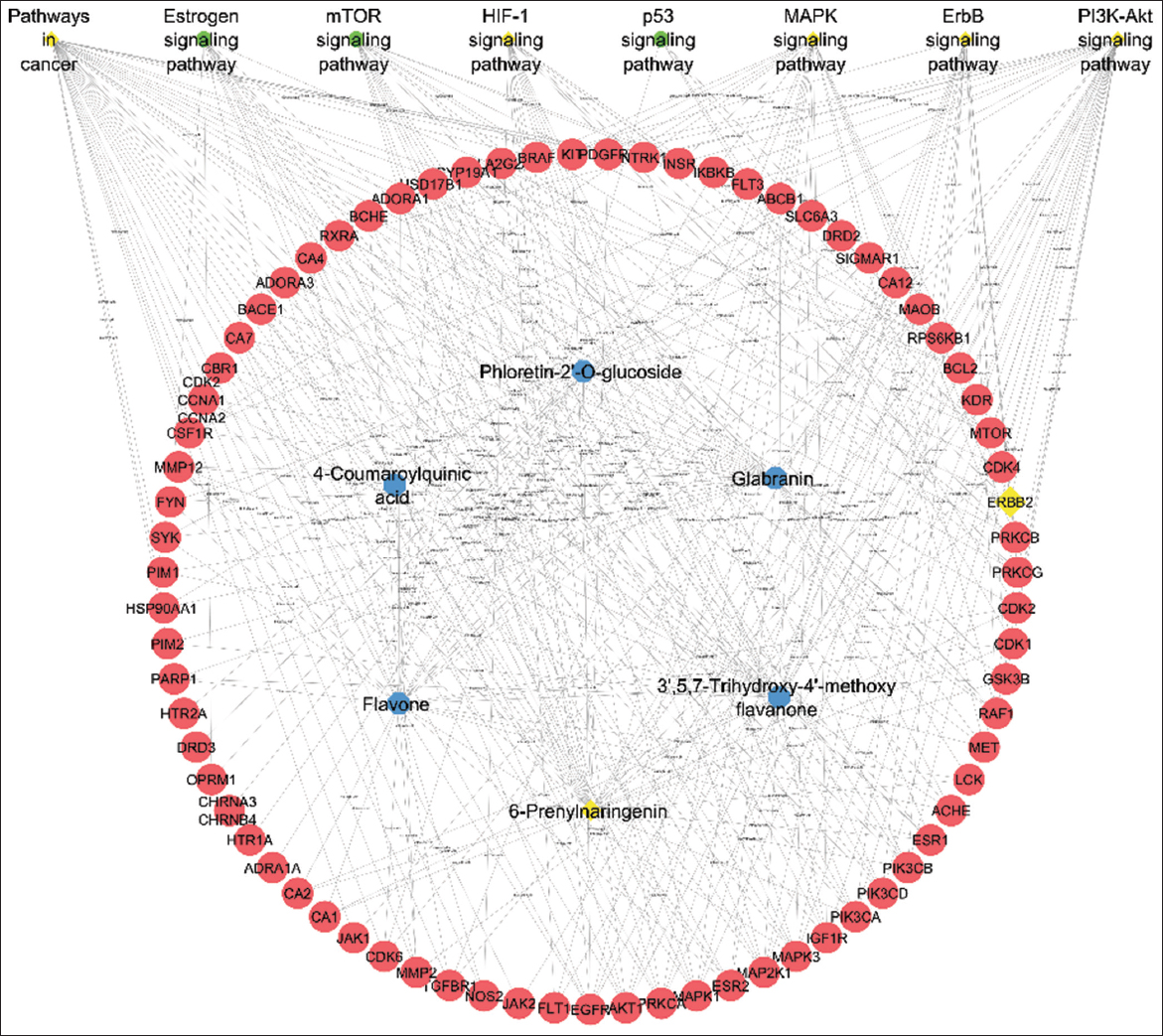 | Figure 4: Network representation of compounds, proteins, and pathway interactions.
[Click here to view] |
Table 5: Pathways associated with breast cancer progression modulated by phytocompounds.
| Pathways | Proteins |
|---|
| Pathways in cancer | MAPK1, BDKRB1, MMP2, RPS6KB1, CDKN1B, FLT3, EGLN3, PDGFRA, CDK4, PDGFRB, MAPK3, PRKCG, BIRC3, PIK3CA, CDK6, CDK2, ERBB2, EGFR, CSF1R, PPARG, KIT, STAT6, MAP2K1, CXCL8, F2, TERT, CASP3, EDNRA, MET, ROCK2, MMP1, GSK3B, NOS2, HSP90AA1, GSTA1, FGFR3, PTK2, ESR2, RET, STAT1, MTOR, EGLN1, PTGS2, CASP7, GSTO1, XIAP, HSP90AB1, MMP9, ABL1, PIM1, HDAC1, AR, PIM2, PIK3CD, JAK2, ALK, BAD, MAPK8, BCL2, GSTP1, ROCK1, FGFR1, AGTR1, RAF1, ESR1, PRKCA, FGFR2, BRAF, RXRA, IKBKB, NTRK1, JAK3, TGFBR1, BDKRB2, AKT1, BIRC2, PRKCB, IGF1R, JAK1, PIK3CB |
| PI3K-Akt signaling pathway | MAPK1, RPS6KB1, CDKN1B, FLT3, PDGFRA, CDK4, PDGFRB, MAPK3, KDR, PIK3CA, CDK6, CDK2, ERBB2, EGFR, NTRK2, FLT1, CSF1R, KIT, NOS3, MAP2K1, INSR, CHRM1, MET, GSK3B, HSP90AA1, FGFR3, PTK2, PDPK1, EPHA2, MTOR, MCL1, HSP90AB1, SYK, PIK3CD, JAK2, BAD, BCL2, FGFR1, CHRM2, RAF1, PRKCA, FGFR2, PIK3CG, RXRA, IKBKB, NTRK1, JAK3, AKT1, ITGB3, IGF1R, JAK1, PIK3CB |
| MAPK signaling pathway | TNFRSF1A, MAPK1, DUSP3, MAPK14, FLT3, CDC25B, PDGFRA, PDGFRB, MAPK3, MAP3K8, PRKCG, KDR, ERBB2, EGFR, NTRK2, FLT1, CSF1R, KIT, DUSP16, MAP2K1, INSR, CASP3, MET, FGFR3, EPHA2, MAP3K5, CACNA1S, MAPKAPK2, PLA2G4A, MAPK8, FGFR1, RAF1, PRKCA, FGFR2, BRAF, STK3, IKBKB, NTRK1, TGFBR1, AKT1, IRAK4, PRKCB, IGF1R |
| HIF-1 signaling pathway | MAPK1, SERPINE1, RPS6KB1, CDKN1B, EGLN3, MAPK3, PRKCG, PIK3CA, ERBB2, EGFR, FLT1, NOS3, MAP2K1, INSR, NOS2, MTOR, EGLN1, PIK3CD, PDK1, BCL2, PRKCA, AKT1, PRKCB, IGF1R, PIK3CB |
| ErbB signaling pathway | MAPK1, RPS6KB1, CDKN1B, MAPK3, PRKCG, PIK3CA, ERBB2, EGFR, MAP2K1, GSK3B, PTK2, MTOR, ABL1, SRC, PIK3CD, BAD, MAPK8, RAF1, PRKCA, BRAF, AKT1, PRKCB, PIK3CB |
| Estrogen signaling pathway | MAPK1, MMP2, MAPK3, PIK3CA, EGFR, NOS3, MAP2K1, PRKCD, HSP90AA1, ESR2, HSP90AB1, MMP9, SRC, PIK3CD, BCL2, OPRM1, RAF1, ESR1, AKT1, PIK3CB |
| mTOR signaling pathway | TNFRSF1A, MAPK1, RPS6KB1, MAPK3, PRKCG, PIK3CA, MAP2K1, INSR, GSK3B, PDPK1, MTOR, PIK3CD, RAF1, PRKCA, BRAF, IKBKB, AKT1, PRKCB, IGF1R, PIK3CB |
| p53 signaling pathway | SERPINE1, CDK4, CDK6, CDK2, CASP3, CDK1, BCL2, CHEK1 |
REFERENCES
1. Cock IE. The medicinal properties and phytochemistry of plants of the genus Terminalia (Combretaceae). Inflammopharmacology. 2015;23:203-29. [CrossRef]
2. Hossain MA, Uddin MS, Shumi W, Shukor NA. Depulping of fruits and soaking the seeds enhances the seed germination and initial growth performance of Terminalia belerica Roxb. Seedlings. Am J Plant Sci. 2014;5(5):714-25. [CrossRef]
3. Kuniyal CP, Purohit V, Butola JS, Sundriyal RC. Seed size correlates seedling emergence in Terminalia bellirica. South Afr J Bot. 2013;87:92-4. [CrossRef]
4. Hegde SN, Lavanya Devi K, Choudhary M, Menon N, Singh G. A comprehensive metabolome profiling of Terminalia chebula, Terminalia bellirica, and Phyllanthus emblica to explore the medicinal potential of triphala. Sci Rep. 2024;14:31635. [CrossRef]
5. Chalise JP, Acharya K, Gurung N, Bhusal RP, Gurung R, Skalko-Basnet N, et al. Antioxidant activity and polyphenol content in edible wild fruits from Nepal. Int J Food Sci Nutr. 2010;61(4):425-32. [CrossRef]
6. Dharmaratne MP, Manoraj A, Thevanesam V, Ekanayake A, Kumar NS, Liyanapathirana V, et al. Terminalia bellirica fruit extracts: In-vitro antibacterial activity against selected multidrug-resistant bacteria, radical scavenging activity and cytotoxicity study on BHK-21 Cells. BMC Complement Altern Med. 2018;18(1):325. [CrossRef]
7. Chang Z, Jian P, Zhang Q, Liang W, Zhou K, Hu Q, et al. Tannins in Terminalia bellirica inhibit hepatocellular carcinoma growth by regulating EGFR-signaling and tumor immunity. Food Funct. 2021;12(8):3720-39. [CrossRef]
8. Pinmai K, Chunlaratthanabhorn S, Ngamkitidechakul C, Soonthornchareon N, Hahnvajanawong C. Synergistic growth inhibitory effects of Phyllanthus emblica and Terminalia bellirica extracts with conventional cytotoxic agents: Doxorubicin and cisplatin against human hepatocellular carcinoma and lung cancer cells. World J Gastroenterol. 2008;14(10):1491-7. [CrossRef]
9. Bakchi B, Krishna AD, Sreecharan E, Ganesh VB, Niharika M, Maharshi S, et al. An overview on applications of Swiss ADME web tool in the design and development of anticancer, antitubercular and antimicrobial agents:A medicinal chemist's perspective. J Mol Struct. 2022;1259:132712. [CrossRef]
10. Ononamadu CJ, Seidel V. Exploring the antidiabetic potential of Salvia officinalis using network pharmacology, molecular docking and ADME/drug-likeness predictions. Plants (Basel). 2024;13:2892. [CrossRef]
11. El Rhabori S, Alaqarbeh M, El Allouche Y, Naanaai L, El Aissouq A, Bouachrine M, et al. Exploring innovative strategies for identifying anti-breast cancer compounds by integrating 2D/3D-QSAR, molecular docking analyses, ADMET predictions, molecular dynamics simulations, and MM-PBSA approaches. J Mol Struct. 2025;1320:139500. [CrossRef]
12. Gupta R, Singh RL, Dwivedi N. In vitro antioxidant activity and GC-MS analysis of the ethanolic extracts of Terminalia bellirica Roxb (baheda). Int J Pharm Pharm Sci. 2016;8:275-82. [CrossRef]
13. Sobeh M, Mahmoud MF, Hasan RA, Abdelfattah MA, Osman S, Rashid HO, et al. Chemical composition, antioxidant and hepatoprotective activities of methanol extracts from leaves of Terminalia bellirica and Terminalia sericea (Combretaceae). PeerJ. 2019;7:e6322. [CrossRef]
14. Ke Z, Wang G, Yang L, Qiu H, Wu H, Du M, et al. Crude terpene glycoside component from radix paeoniae rubra protects against isoproterenol-induced myocardial ischemic injury via activation of the PI3K/AKT/MTOR signalling pathway. J Ethnopharmacol. 2017;206:160-9. [CrossRef]
15. Vago R, Bettiga A, Salonia A, Ciuffreda P, Ottria R. Development of new inhibitors for N-acylethanolamine-hydrolyzing acid amidase as promising tool against bladder cancer. Bioorg Med Chem. 2017;25:1242-9. [CrossRef]
16. Elizabeth LA, Bupesh G, Susshmitha R. In vitro antioxidant efficacy of Terminalia bellirica seed extract against free radicals. Int J Pharm Sci Res. 2017;8:4659-65. https://doi.org/10.13040/ijpsr.0975-8232.8(11).4659-65 [CrossRef]
17. Patil SB, Kuvalekar MB, Yaraguppi DA, Prasanth DS, Halkavatagi SG, Tennalli GB, et al. Exploring the efficacy of Benincasa hispida extract on obesity linked inflammatory bowel disease by integrating computational analysis and experimental validations. Sci Rep. 2025;15(1):14426. [CrossRef]
18. Channalli SR, Pattanashetti L, Patil SB, Yaraguppi DA, Prasanth DS, Halkavatagi SG, et al. Deciphering the multitarget neuroprotective potential of Ficus microcarpa L. f. Leaf extract: Insights from phytochemical, computational, and experimental approaches. Chem Biodivers. 2025;23:e02568. [CrossRef]
19. Banerjee P, Kemmler E, Dunkel M, Preissner R. ProTox 3.0:A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024;52(W1):W513-20. [CrossRef]
20. Mane RR, Yaraguppi DA, Bagewadi ZK, Kamanna K. Organocatalysed synthesis of N-(4-oxo-2-phenyl-1,2-dihydroquinazolin-3(4 h)-yl)isonicotinamide: Computational, electrochemical, drug-likeness and antimicrobial studies. 3 Biotech. 2025;15(1):30. [CrossRef]
21. Yan L, Zhang Z, Liu Y, Ren S, Zhu Z, Wei L, et al. Anticancer activity of erianin: Cancer-specific target prediction based on network pharmacology. Front Mol Biosci. 2022;9:862932. [CrossRef]
22. Huang F, Fu M, Li J, Chen L, Feng K, Huang T, et al. Analysis and prediction of protein stability based on interaction network, gene ontology, and KEGG pathway enrichment scores. Biochim Biophys Acta Proteins Proteom. 2023;1871:140889. [CrossRef]
23. Piñero J, Saüch J, Sanz F, Furlong LI. The DisGeNET cytoscape app: Exploring and visualizing disease genomics data. Comput Struct Biotechnol J. 2021;19:2960-7. [CrossRef]
24. Gummagol NB, Yaraguppi DA, Patil SB, Patil PS, Patil NR, Ayachit NH. Exploring the anticancer potential of novel chalcone derivatives: Synthesis, characterization, computational analysis, and biological evaluation against breast cancer. J Mol Struct. 2025;1320:139586. [CrossRef]
25. Hebasur RK, Koppal VV, Yaraguppi DA, Gummagol NB, Kusanur R, Patil NR. A comprehensive evaluation of a chalcone derivative: Structural, spectroscopic, computational, electrochemical, and pharmacological perspectives. Photochem. 2025;5(3):20. [CrossRef]
26. Sehgal R, Sharma AK, Singh BJ, Saini RV, Saini AK, Beniwal V. Augmenting the antioxidant, anti-bacterial and anti-carcinogenic potential of Terminalia chebula and Terminalia bellirica after tannin acyl hydrolase mediated biotransformation. Biocatal Agric Biotechnol. 2024;56:103045. [CrossRef]
27. Mane R, Yaraguppi DA, Chandrakala KB, Kamanna K. Synthesis, computational, anticancer, and electrochemical behavior studies of 3-methyl-4-(hetero)aryl methylene isoxazole-5(4h)-one. ChemistrySelect. 2024;9:e202401158. [CrossRef]
28. Wan H, Williams R, Doherty P, Williams DF. A study of the reproducibility of the MTT test. J Mater Sci Mater Med. 1994;5(3):154-9. [CrossRef]
29. Umesh Kanna S, Parthiban KT, Senthilraja K, Venkatesan S, Udhaya Nandhini D, Mohan Kumar S, et al. Genetic diversity and structure of Terminalia bellirica (gaertn. Roxb.) Population in India as revealed by genetic analysis. Plants (Basel). 2024;13:470. [CrossRef]
30. Srinivasan M, Gangurde A, Chandane AY, Tagalpallewar A, Pawar A, Baheti AM. Integrating network pharmacology and in silico analysis deciphers Withaferin-A's anti-breast cancer potential via hedgehog pathway and target network interplay. Br Bioinform. 2024;25:bbae032. [CrossRef]
31. Zemnou CT. Network pharmacology, molecular docking, and molecular dynamics simulation revealed the molecular targets and potential mechanism of Nauclea latifolia in the treatment of breast cancer. Chem Biodivers. 2024;22:e202402423. [CrossRef]
32. Baothman OA, Islam MR. Exploring breast cancer treatment paradigms: Innovative design, molecular docking and dynamic simulation of LOXL2 inhibitors. Mol Simul. 2024;50:631-43. [CrossRef]
33. Yang Z, Wang Y, Huang S, Geng Y, Yang Z, Yang Z. Identification of potential anti-tumor targets and mechanisms of huachansu injection using network pharmacology and cytological experiments in breast cancer. PLoS One. 2024;19:e0303650. [CrossRef]
34. El Rhabori S, Alaqarbeh M, El Aissouq A, Bouachrine M, Chtita S, Khalil F. Design, 3D-QSAR, molecular docking, ADMET, molecular dynamics and MM-PBSA simulations for new anti-breast cancer agents. Chem Phys Impact. 2024;8:100455. [CrossRef]
35. Zochedh A, Chandran K, Priya M, Sultan AB, Kathiresan T. DFT simulation of berberine chloride with spectroscopic characterization - biological activity and molecular docking against breast cancer. Polycycl Aromat Compd. 2024;44:1556-80. [CrossRef]
36. Patil PV, Nerlekar NA, Kuldeep AR, Patil PP, Dandge PB, Dongale TD, et al. Terminalia bellirica (Gaertn.) Roxb. Extract-mediated green synthesis of magnesium oxide nanoparticles for multifunctional applications. Plant Nano Biol. 2024;8:100069. [CrossRef]
37. Venturelli S, Niessner H, Sinnberg T, Berger A, Burkard M, Urmann C, et al. 6- and 8-prenylnaringenin, novel natural histone deacetylase inhibitors found in hops, exert antitumor activity on melanoma cells. Cell Physiol Biochem. 2018;51(2):543-56. [CrossRef]