
1. INTRODUCTION
The second most frequent malignancy among women is breast cancer, which presents a global challenge due to its intricate, multi-step nature involving various cell types [1]. Early diagnosis remains an important strategy for the prevention of breast cancer. In certain developed countries, the 5-year relative survival rate for patients with breast cancer surpasses 80%, which is primarily attributed to early detection and prompt treatment [2]. Over time, many cancers gain resistance to current therapies, hence lowering the effectiveness of treatments such as chemotherapy and targeted therapy, among others. Tumors can gain resistance through mutation or adaptation, and this is where the potency of the developed drugs fades out [3]. There are promising benefits of anticancer molecules derived from biological sources, such as plants, marine organisms, fungi, and microorganisms, which are now under study against traditional synthetic drugs.
On the other hand, many natural products function as selective toxins. They selectively kill cancer cells with little harm to healthy tissues, which may minimize the side effects common to traditional chemotherapy [4]. Over the past decade, significant progress has been made in understanding breast cancer and devising preventive measures. The identification of breast cancer stem cells has provided insights into the pathophysiology and mechanisms of drug resistance in tumors. In addition, numerous genes associated with breast cancer have been pinpointed [5]. This heightened understanding has paved the way for advancements in preventive strategies.
The past 10 years have witnessed substantial strides in both comprehending breast cancer and in formulating preventive approaches. Breast cancer stem cell research has provided insight into the pathophysiology and underlying causes of tumor drug resistance, and the identification of several genes linked to breast cancer has contributed to our knowledge [5]. At present, more pharmaceutical options are available for the chemoprevention of breast cancer, and recent developments in biological prevention aim to enhance the quality of life of patients. Approximately 1.5 million women worldwide, constituting 25% of all cancer patients, receive a breast cancer diagnosis each year [6]. The development and maturation of human breast alveolar glands relies on progesterone, which collaborates with estradiol rather than functioning independently as a mitogenic hormone. This collaboration is essential for preparing the mammary gland to respond to mitogens such as prolactin, glucocorticoids, and growth hormones [7].
Progesterone receptors (PGRs), members of the ligand-activated transcription factor family, belong to the nuclear receptor family of the steroid hormone receptor (SR) subfamily. The full-length receptor is denoted as PGR-B, whereas the N-terminally truncated variant, PGR-A, is produced from the same gene utilizing alternative translational start sites and lacks the initial 164 amino acids present in PGR-B. Two distinct (isomer-specific) promoters independently regulate PGR. Both PGR-A and PGR-B can bind to DNA as homo- (A: A or B: B) or heterodimers (A: B) at progesterone response sites [6]. They may also interact with other transcription factors, including specificity protein 1 (SP1), activator protein 1, and signal transducers and activators of transcription (STATs) [7,8]. Notably, both PGR-A and PGR-B exhibit ligand-dependent and ligand-independent actions, enabling them to regulate similar or distinct sets of target genes based on the expressed isoform.
PGR belong to the family of steroid nuclear receptors and are recognized as ligand-dependent transcription factors. It consists of an inherently disordered protein-containing amino-terminal domain, ligand-binding domain (LBD) at the C-terminus, and DNA-binding domain (DBD) in the central region [9]. The transcriptional activation domains or functions (AFs) of the PGR have two interfaces for interactions with co-regulatory proteins. AF2 is located in the LBD, whereas AF1 is located in the N-terminal domain. There are two primary isoforms of the PGR, PGR-A and PGR-B, both of which originate from the same gene on 11q22-q23 using different promoters. These isoforms exhibit distinct biological and transcriptional characteristics as ligand-activated transcription factors [10,11]. PGR-B functions as a fully functional receptor, whereas PGR-A is a truncated form of PGR-B that lacks 164 amino acids from the N-terminus of the protein. In progestin-independent contexts, PGR-A is more active than PGR-B [12] and is primarily localized in the nucleus, failing to mediate direct transcriptional events or rapid cytoplasmic changes [13].
PGR regulates estrogen receptors (ER) in breast cancer, serving as an ER-target gene whose expression is dependent on estrogen. Progesterone, a 21-carbon steroid hormone, binds to PGRs and plays a crucial role in the female menstrual cycle, pregnancy, and embryogenesis in various species [14]. PGR is a valuable prognostic biomarker, particularly in hormone-positive breast cancer. In cancers classified as luminal A, which are associated with a more favorable prognosis than luminal B, a less favorable subtype, heightened PGR expression is frequently observed. PGR signaling plays a crucial role in ductal elongation and side branching in response to increased estrogen levels in the epithelial compartment [15]. During early pregnancy, PGR signaling may contribute to the sudden expansion of the epithelial compartment. Progesterone is essential for alveolar differentiation in the middle to late stages of pregnancy, but at term, it transforms into an inhibitor of terminal differentiation, necessitating its removal to facilitate lactation [16].
The progression and formation of breast cancer, along with the healthy development of mammary glands, relies on the PGR SR. PGR is a routinely employed biomarker during breast cancer diagnosis and plays a role in molecular subtyping, thereby influencing therapy selection. However, it is crucial to elucidate their intricate roles under varying conditions. In clinical settings, PGR-positive breast cancers exhibit a higher endocrine response than PGR-negative breast cancers. Nonetheless, its utility as a consistent predictor of endocrine therapy outcomes is sporadic.
The anticancer properties of lipopeptides derived from Bacillus subtilis are both effective and productive [17]. Various lipopeptides produced by B. subtilis, such as surfactin, iturin, bacilomycin D, and fengycina, have demonstrated potent anticancer activity in diverse cancer cell lines. Surfactin, when increased in human colon cancer cell lines, has been found to impede cancer metastasis [18]. Reports indicate that it exhibits cytotoxic activity in cervical cancer, induces apoptosis in oral cancer [19], and exerts a cytotoxic effect on pancreatic cancer. In addition, it has been proven effective in eliminating human breast carcinoma cell lines [10]. Iturin, another lipopeptide, hinders cell proliferation and induces apoptosis in breast cancer cell lines MCF-7 and MDA-MB-231 [11], colon adenocarcinoma cell lines HCT-15 and A549, and renal carcinoma cell lines A498 [20]. HCC cells exhibited potent anticancer activity against B. subtilis iturin A, both in vitro and in vivo. Iturin A greatly inhibited the growth of tumor cells in vitro and induced autophagy, paraptosis, and apoptosis. The inhibitory action was dose-dependent and could rapidly penetrate the cancer cells [21].
Network-based research and pharmacology represent two of the most recent areas of investigation, offering potential strategies to streamline the drug development process economically [22]. Network pharmacology is currently being explored as an emerging paradigm in drug development, leveraging concepts from systems biology and network theory to yield a comprehensive or partial understanding of pharmacological data [23]. It systematically incorporates data to delineate interactions between medications and their targets, encompassing various procedures such as active component screening, enrichment analysis, target identification, and utilization of disease target databases. The proposed bottom-up approach aims to expedite the discovery of phytopharmaceuticals by generating testable experimental hypotheses regarding potential mechanisms [24].
The molecular docking technique evaluates the binding surface and interaction forces between the receptor and ligand to estimate the binding mechanism and affinity of the receptor-ligand complex [25]. MD simulation can assess the structural stability of the receptor and ligand, as well as the kinetics of the receptor-ligand interaction, based on an appropriate force field [26]. Numerous biological peptides and proteins have been documented in the literature for the treatment of various cancers; however, the quest for novel biologically active proteins targeting different cancer types persists. This impetus fuels research to explore the anticancer potential of recombinant iturin A. Our study systematically assessed the cytotoxic effect of recombinant iturin A on breast cancer MCF-7 cell lines and normal mammalian L929 cell lines using the MTT assay, a pivotal tool for evaluating the anticancer potential of molecules. In the present study, pathways related to iturin A in breast cancer treatment were extensively explored using a combination of network pharmacology, molecular docking, MD simulation, and in vitro studies.
2. METHODS
2.1. Chemicals and Cell Lines
Different chemicals used for the present study are yellow tetrazolium MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide, Dimethyl Sulfoxide (DMSO), Dulbecco’s Modified Eagle Medium, and trypsin. The chemicals used for the cytotoxicity assay were procured from Gibco (USA), Invitrogen (USA), and Thermo Fisher Scientific (USA). The cell lines employed in this study included breast cancer cells (MCF-7) and normal mammalian cells (L929), which were procured from the National Center for Cell Science (NCCS), Pune, Maharashtra, India.
2.2. Molecular Properties and Drug-Likeness of Iturin A
The molecular properties and drug-likeness of the compound were evaluated using the Molsoft Tool, a computational platform designed for assessing physicochemical characteristics and drug-likeness potential. The input for the analysis included the molecular structure of the compound, represented by its molecular formula, stereochemistry, and associated features. The tool utilized advanced algorithms to compute essential molecular descriptors such as molecular weight, hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), topological polar surface area (MolPSA), molecular volume (MolVol), and lipophilicity (MolLogP). Solubility (MolLogS) and the pKa and pKb values of the most basic and acidic groups were also determined to provide insights into the compound’s physicochemical behavior in different biological environments. The computational analysis further evaluated the compound’s potential to cross the blood-brain barrier (BBB), quantified by a BBB Score (ranging from 6 for high penetration to 0 for low penetration). The number of stereocenters in the molecule was identified, reflecting its complexity. In addition, the drug-likeness model score was calculated to assess the compound’s suitability as a potential drug candidate, considering factors such as molecular weight, HBA, HBD, and MolLogP, compared to established thresholds [27].
2.3. Target Identification
Canonical SMILES were analyzed for target prediction using Swiss Target Prediction (
2.4. Pathway and Network Analysis
The STRING database (
2.5. Molecular Docking of Compound and Target
PPI network chemicals and breast cancer-related targets for molecular docking. The interactions between the suspected active compounds and significant targets were investigated using the software programs AutoDock Vina, Discovery Studio 4.5 Client, and PyMOL. The binding affinity and mode of action of a small-molecule ligand with a target protein were predicted using molecular docking. Through the activation of CREB/CREM/ATF-1 and overexpression of PKA/cAMP, the PGR contributes to cell proliferation. In addition, when PR is activated, the Wnt/β-catenin pathway is upregulated, which promotes cell growth and carcinogenesis by activating MAPK/SRC through overexpression of the transcription factor Sp1 [34]. Based on network pharmacology and a literature survey, the PGR was selected for docking and simulation studies. The 3D structures of iturin A were sourced from ChEBI database (
2.6. Molecular Dynamics (MD) Simulation
MD simulations play a pivotal role in assessing the stability and compactness of the docked complexes. In the context of our study, an MD simulation was conducted using Gromacs-2019 for the selected protein-ligand pairing. To prepare the system, they were minimized for 1500 steps using the steepest descent method. Subsequently, the complex structures were solvated in a cubic periodic box (0.5 nm) using a simple point charge water model. Maintaining a salt content of 0.15 M, appropriate quantities of Na+ and Cl− counterions were added. To properly equilibrate the modeled systems, systems were equilibrated under NVT and NPT conditions while slowly relaxing the position restraints applied to the atoms. The resulting structure from the ensemble was subjected to a production run for a chemical time of 200 ns. Trajectory analysis was performed using the analysis modules offered by the Gromacs simulation suite, focusing on the backbone root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (RG), solvent accessible surface area (SASA), and H-bond, as detailed in our previous studies [37].
2.7. Principal Component Analysis (PCA) and Gibbs Free Energy (GFE) Calculation
PCA and GFE were employed as analytical tools to comprehend structural changes in our study. PCA provides insights into the overall motion and variability of atomic coordinates, while GFE offers a thermodynamic perspective, helping to assess the stability of different protein conformations. Together, these analyses contribute to a comprehensive understanding of the dynamic behavior and energetic landscape of the system. PCA was employed to capture pronounced fluctuations in protein residues, as outlined [38]. Simultaneously, the variability in GFE values was considered when assessing the stability levels of protein molecules, a crucial factor for their proper functional performance, as highlighted by the studies conducted by Rajendran and Sethumadhavan [39]. The calculation of GFE is instrumental in elucidating the thermodynamic properties underlying the formation of antibody-antigen complexes and PPI [40]. We computed the covariance matrix by exclusively considering the Cα coordinates. Subsequent diagonalization facilitated the derivation of the Eigenvectors and Eigenvalues. PCA was then performed using a trajectory comprising 2000 snapshots, and the first two principal components (PC1 and PC2) were extracted for subsequent analysis. The free-energy landscape was also generated using methodologies outlined in prior studies by Sun et al. [41] and Martis and Coutinho [42].
2.8. Evaluation of Cytotoxicity
Recombinant iturin A, previously produced and purified as detailed in our earlier publication [43], was subjected to cytotoxicity assessment using the methodology outlined by Shettar et al. [44]. The in vitro 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay protocol was followed. The effect of recombinant iturin A on cell viability was examined in breast cancer MCF-7 and normal mammalian L929 cell lines with an effective concentration range of 3.125–100 μg/mL. It was compared with the standard drug doxorubicin with the concentration range of 3.125–100 μg/mL. Each treatment was conducted in triplicate, with doxorubicin serving as a standard drug [45].
In a 96-well microplate, 5000 cells/well were seeded for a 24-h period. Subsequently, cells were treated with varying concentrations of iturin A and incubated for 24 h. After repetitive washing with phosphate-buffered saline, 100 μL of medium containing 10% MTT reagent was then added to each well to get a final concentration of 0.5 mg/mL, and the cells were incubated for an additional 5 h. The formazan crystals obtained were solubilized in DMSO, and the absorbance was measured at 570 nm. Untreated cells served as controls. The percentage of cell viability in iturin A-treated cells was calculated and presented as a plot of cell viability % versus iturin A concentration.
2.9. Apoptosis Based on Flow Cytometry
For cancer, the inhibition of apoptosis is one of the fundamental requirements for the initiation and development of the disease and most frequently results in the failure of drug-based treatments due to resistance. Here, we investigated the cytotoxic activity of the recombinant protein using flow cytometry combined with annexin V-FITC/propidium iodide (PI) double staining, which is one of the standard methodologies applied for the quantification of a population of apoptotic cells. Annexin V-FITC staining was used to evaluate the translocation of phosphatidylserine from the inner leaflet to the outer leaflet of the plasma membrane during early apoptosis. Given that the integrity of the cell membrane decreases as apoptosis continues, PI can penetrate and stain DNA inside the nucleus. Utilizing recombinant iturin at its IC50 concentration against MCF-7 cells, our study was evaluated for a period of 24 h following the procedures in the assay kit. We then used FlowJoX 10.0.7 software to analyze the identification and measurement of early and late apoptotic events after incubation.
3. RESULTS
3.1. Selection and Preparation of Iturin A
The absorption, distribution, metabolism, excretion, and toxicity analysis of iturin A [Table 1] indicated no adverse effects for the screened substances. Positive outcomes were observed for P-glycoprotein substrates, Caco-2 permeability, and BBB penetration, underscoring the potential of these bioactive substances as innovative therapeutic agents. Notably, in assessments across various toxicity categories, all predicted chemicals were identified as non-toxic and exhibited no toxic behavior [46].
Table 1: Absorption, distribution, metabolism, excretion, and toxicity profiling of the iturin A.
| Parameters | Values |
|---|---|
| Blood-brain barrier | 0.9521 |
| Caco-2 permeability | 0.8661 |
| Pgp-inhibitor | 0.9671 |
| Pgp-substrate | 0.6635 |
| CYP1A2 inhibitor | 0 |
| CYP1A2 substrate | 0 |
| CYP2C19 inhibitor | 0.004 |
| CYP2C19 substrate | 0.011 |
| CYP2C9 inhibitor | 0.021 |
| CYP2C9 substrate | 0.026 |
| CYP3A4 inhibitor | 0.001 |
| CYP3A4 substrate | 0.001 |
| Acute toxicity | 0 |
| Ames toxicity | 0.02 |
| Solubility | 1.02e-05 mg/mL; 9.75e-09 moL/L |
| Log Kp (skin permeation) | −13.97 cm/s |
| Log Po/w (iLOGP) | 0.62 |
| pKa | 9.89 |
| pKb | 4.11 |
3.2. Gene set Enrichment and Network Pharmacology
3.2.1. Target identification
The canonical SMILES structure of Iturin A was analyzed using SwissTargetPrediction to identify potential therapeutic targets. This analysis revealed 56 compound-related targets, which were further cross-referenced with known therapeutic drug molecules to validate their relevance. Simultaneously, a search of the GeneCards database identified 18,337 target proteins associated with breast cancer. These targets were meticulously analyzed to identify intersections between Iturin A-related genes and breast cancer-related genes. Using a Venn diagram approach and shown in Figure 1, 53 combined genes were identified, representing the shared genetic landscape between the compound’s activity and the disease. These genes serve as the basis for further exploration into the compound’s mechanism of action and therapeutic potential.
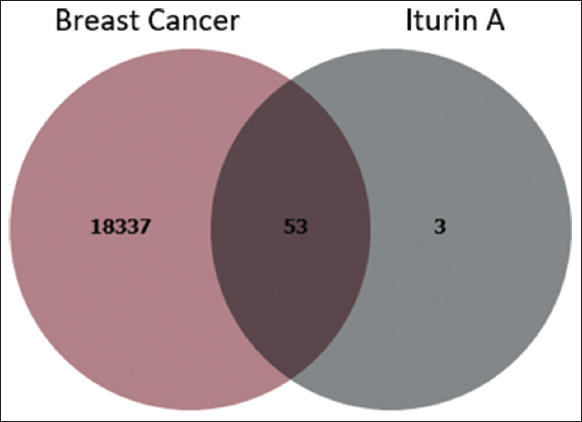 | Figure 1: Venn diagram of breast cancer and target compound. [Click here to view] |
3.2.2. Pathway and network analysis
The 56 identified compound targets were uploaded into the STRING database to map PPI [Figure 2] and to perform molecular pathway analysis. In parallel, data from the KEGG database and an extensive literature review were used to identify signaling pathways relevant to breast cancer. Key pathways and their associated protein targets are HIF-1 signaling pathway: Includes MAPK1, CDKN1B, PRKCG, PIK3CA, STAT3, MTOR, PIK3CD, PRKCA, AKT1, and PRKCB. This pathway plays a critical role in cellular adaptation to hypoxia, a common feature in breast cancer progression. VEGF signaling pathway: Involves MAPK1, PRKCG, PIK3CA, SRC, PIK3CD, PRKCA, AKT1, and PRKCB. This pathway regulates angiogenesis, which is essential for tumor growth and metastasis. Estrogen signaling pathway: Encompasses MAPK1, CTSD, PIK3CA, PGR, SRC, PIK3CD, GRB2, OPRM1, and AKT1. The pathway is crucial in hormone-dependent breast cancers, highlighting the interplay between Iturin A and hormonal regulation. mTOR signaling pathway: Targets MAPK1, PRKCG, PIK3CA, MTOR, PIK3CD, GRB2, PRKCA, AKT1, and PRKCB, which are vital in cell growth, proliferation, and survival. PI3K-Akt signaling pathway: Involves MAPK1, CDKN1B, ITGAV, PIK3CA, F2R, MTOR, PIK3CD, GRB2, PRKCA, AKT1, and PRLR. This pathway is frequently dysregulated in breast cancer, leading to uncontrolled cell survival and growth. Breast cancer pathway: Includes MAPK1, PIK3CA, PGR, MTOR, PIK3CD, GRB2, and AKT1, directly correlating with known genetic markers and therapeutic targets for the disease. JAK-STAT signaling pathway involves PIK3CA, STAT3, MTOR, PIK3CD, GRB2, AKT1, and PRLR. This pathway mediates inflammatory and growth signals that contribute to cancer progression. TNF signaling pathway: Targets MAPK1, IL1B, PIK3CA, PIK3CD, and AKT1, involved in inflammation and apoptosis regulation. NF-kappa B signaling pathway: Includes PRKCQ, IL1B, and PRKCB, which modulates inflammatory responses and cell survival mechanisms, commonly activated in cancer [Table 2]. The relationships among compounds, target proteins, and pathways were visualized using Cytoscape v3.6.1. The network visualization employed color coding and variable node sizes, where node size represented edge count (number of connections). Nodes having high edge counts, or degree centrality, act as key hubs in a network. These nodes are pivotal for interconnection between various nodes and for information flow within the network. A high edge count usually correlates with more interactions for a node, thereby increasing the influence that it holds in controlling paths activated within the network and improving the effectiveness of communication along those paths while maintaining the structure of the network. Such connected nodes are likely to represent the most important biological targets, main regulators, or central molecules in biochemical and pharmacological networks. Through combining genes, the PGR was selected, which emerged as a highly significant node due to its strong connections to iturin A, its integration within the estrogen signaling pathway, and its established role in breast cancer progression. The PGR node was visually highlighted in sky blue in Figure 3, underscoring its importance. Furthermore, the identified pathways demonstrate the multifaceted role of iturin A in targeting critical biological processes such as hormone regulation, cell growth, angiogenesis, and inflammation – all of which are implicated in breast cancer development and progression. This network pharmacology approach highlights the potential of iturin A as a multi-target therapeutic agent, particularly for hormone-responsive breast cancer subtypes. The integration of computational target prediction, pathway enrichment analysis, and network visualization provides compelling evidence for the potential therapeutic role of iturin A in breast cancer treatment. The identified PGR protein and its association with the estrogen signaling pathway and breast cancer highlight a promising avenue for further experimental validation. Future molecular docking studies should focus on experimental verification of these interactions between PGR and iturin A and exploring the pharmacokinetic and pharmacodynamic properties to assess its viability as a drug candidate.
 | Figure 2: Probable protein-protein interaction network. [Click here to view] |
Table 2: Pathways associated with breast cancer progression modulated by phytocompounds.
| Pathways | Proteins |
|---|---|
| HIF-1 signaling pathway | MAPK1, CDKN1B, PRKCG, PIK3CA, STAT3, MTOR, PIK3CD, PRKCA, AKT1, and PRKCB |
| VEGF signaling pathway | MAPK1, PRKCG, PIK3CA, SRC, PIK3CD, PRKCA, AKT1, and PRKCB |
| Estrogen signaling pathway | MAPK1, CTSD, PIK3CA, PGR, SRC, PIK3CD, GRB2, OPRM1, and AKT1 |
| Mtor signaling pathway | MAPK1, PRKCG, PIK3CA, MTOR, PIK3CD, GRB2, PRKCA, AKT1, and PRKCB |
| PI3K-Akt signaling pathway | MAPK1, CDKN1B, ITGAV, PIK3CA, F2R, MTOR, PIK3CD, GRB2, PRKCA, AKT1, and PRLR |
| Breast cancer | MAPK1, PIK3CA, PGR, MTOR, PIK3CD, GRB2, and AKT1 |
| JAK-STAT signaling pathway | PIK3CA, STAT3, MTOR, PIK3CD, GRB2, AKT1, and PRLR |
| TNF signaling pathway | MAPK1, IL1B, PIK3CA, PIK3CD, and AKT1 |
| NF-kappa B signaling pathway | PRKCQ, IL1B, and PRKCB |
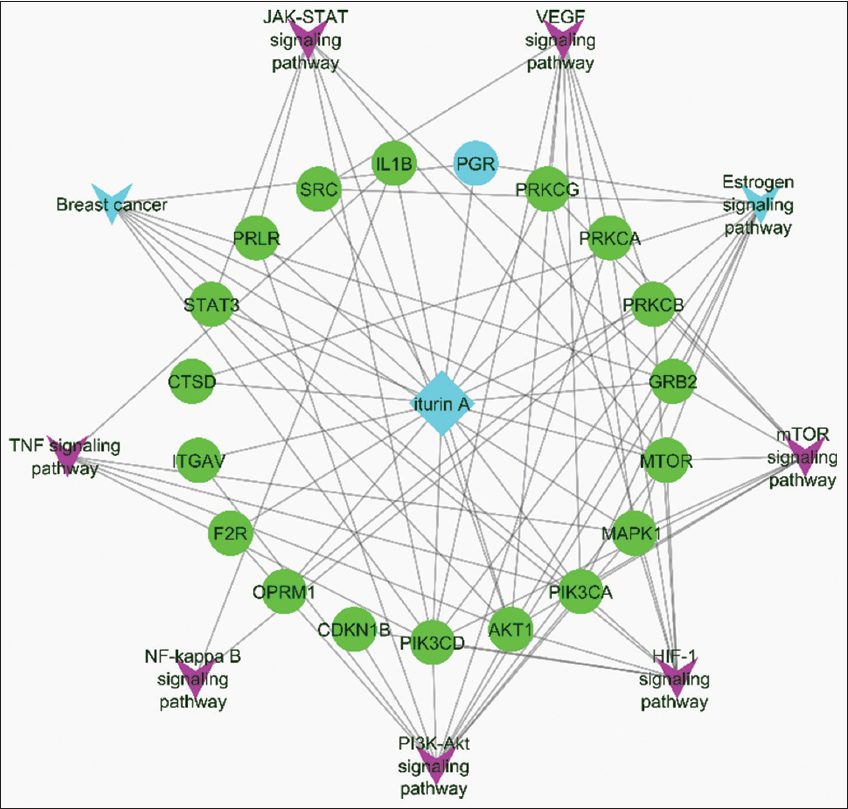 | Figure 3: Network representation of compounds, proteins, and pathway interactions. [Click here to view] |
3.3. Protein Preparation
The crystal structure of the PGR, identified as the target protein, was retrieved from the PDB using PDB ID 4OAR [Figure 4]. This structural foundation forms the basis for extensive investigations of the docking process. Subsequent target docking was performed using AutoDock Vina, with the PGR docking grids set at X: 16.201750, Y: 20.216750, and Z: 19.193750 from the center, and exhaustiveness of 8. The results of docking research revealed a notable binding affinity between the active and core targets.
 | Figure 4: Three-dimensional (3D) predicted structure of target protein. [Click here to view] |
3.4. Molecular Docking
The crystal structure of the PGR, identified as the target protein, was retrieved from the PDB using PDB ID 4OAR. This structural foundation formed the basis for extensive investigations into the docking process. Primary interactions between the ligand and target proteins involve hydrophilic interactions and hydrogen bonding, leading to binding energy, these types of interactions happen due to polar and non-polar amino acids present in protein. Remarkably, the PGR demonstrated a notable binding affinity for iturin A, establishing two H-bond interactions with specific amino acid residues, including GLN:916 (H1), GLN:720 (H2), GLU:723, LEU:726, TRP:755, and LEU:715. This interaction resulted in a binding energy of −5.5 Kcal/moL, as outlined in Table 3. The detailed interactions, which were generated using the Discovery Studio visualizer, are tabulated and visually represented in Figure 5.
Table 3: Binding energies of the receptor-ligand complexes.
| Protein bank Id | Compound name | Binding affinity (kcal/moL) | Root mean square deviation | No. Hydrogen bonds | Interacting residues |
|---|---|---|---|---|---|
| 4OAR | Iturin A | −5.5 | 0.000 | 02 | GLN: 916(H1), GLN: 720(H2) |
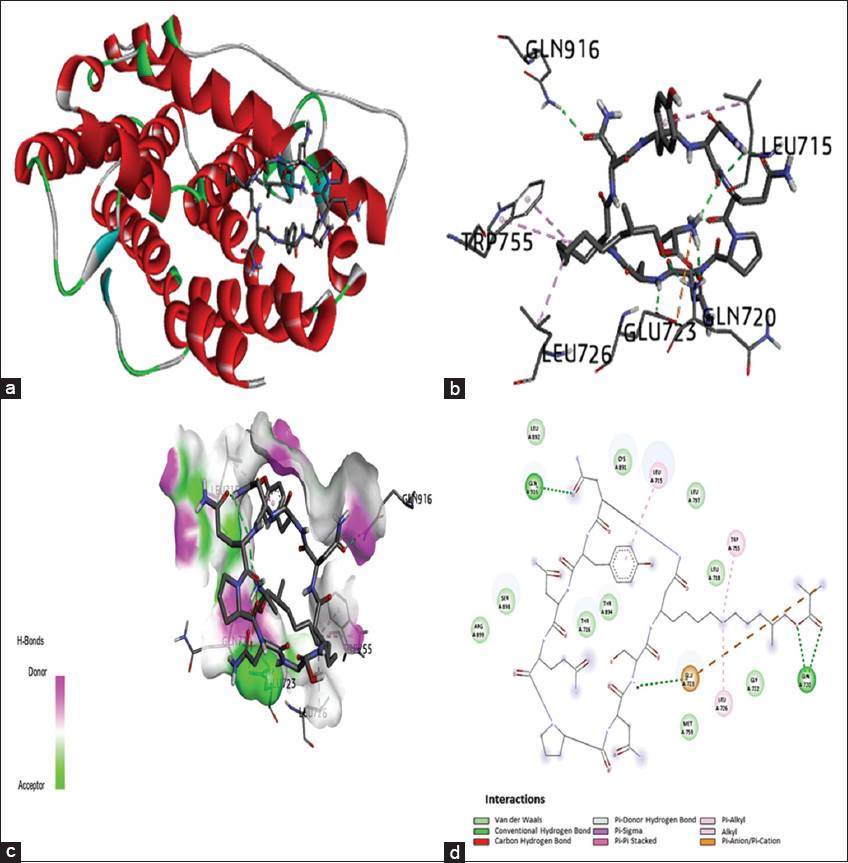 | Figure 5: Molecular docking of the progesterone receptor (4OAR)-binding domain complexed with iturin A shows a 3D model of the interactions, 2D interaction patterns, and H-bond interaction. (a) 3D representation of the receptor-ligand complex. (b) Ligand interaction with amino acids of the protein in 3D representation; (c) H-bond interaction cavity occupancy with a depiction of hydrogen bond acceptor and donor. (d) 2D interaction diagram of the complex showing different types of interactions between the compound and amino acid residues of the protein. [Click here to view] |
3.5. MD Analysis (RMSD, RMSF, RG, SASA, H-bond, and MMPBSA)
MD simulation examined a molecule featuring specific ligands identified through docking. This simulation was extended over a duration of 200 nanoseconds (ns) to enhance our comprehension of the stability exhibited by the aforementioned protein-ligand complexes. This analysis is crucial for understanding the differences between the two confirmations. Notably, as the RMSD increased, the level of divergence increased. The RMSD values, calculated over a simulation timescale of 200 ns using the RMS tool, are presented in the plot below for the protein-ligand structure within the PGR-binding domain (4OAR). The average RMSDs for complexes with iturin A ranged from 0.15 nm to 0.2 nm over the simulation period of 0–200 ns [Figure 6a]. These RMSD values signify the relative stability of the chemical compounds during simulation, suggesting a stable and well-interacting complex between the protein and ligand. RMSF analysis reveals which amino acids in the protein cause the most vibrations. RMSF was calculated by performing a simulation with a duration of 0–200ns. The results of the RMSF for iturin A complex with the PGR (4OAR)-binding domain are depicted in Figure 6b. To assess the folding and unfolding dynamics, RG values were analyzed across a simulated timescale of 0–200 ns for the PGR (4OAR) binding domain Complexed with iturin A. Figure 6c illustrates the RG results for the APO and ligand complexes. In this investigation, the compactness and stability of the ligand were highlighted by the radius of the gyration plot, which revealed values consistently at or below 1.9 nm. As shown in Figure 6d, which encompasses SASA values ranging from 0 to 200, SASA represents the portion of a protein’s surface exposed to the solvent and serves as an indicator of protein stability. The analysis revealed the stable nature of SASA, with no substantial variations in solvent accessibility observed throughout the simulation. Specifically, the solvent accessibility values remained consistently within the 135–140 nm2 range, indicating a consistent and stable protein structure. The formation of H-bonds facilitates the stabilization of protein-ligand complexes. Our study aligns with this principle, as molecular docking analysis indicated the creation of H-bonds, a finding substantiated by simulation analysis. Figure 6e visually represents the iturin A H-bond complex with the progesterone receptor (4OAR)-binding domain.
 | Figure 6: (a) Root mean square deviation of backbone atoms of the progesterone receptor (4OAR)-binding domain complexed with iturin A. (b) root mean square fluctuation (RMSF) of the progesterone receptor (4OAR) binding domain complexed with iturin A. (c) Radius of gyration (RG) of progesterone receptor (4OAR) binding domain complexed with iturin A. (d) SASA of backbone atoms with the progesterone receptor (4OAR)-binding domain complexed with iturin A. (e) H-bond interactions between 4OAR and iturin A. (f) MMPBSA of progesterone receptor (4OAR) binding domain complexed with iturin A. [Click here to view] |
Culminating in the fact that MD simulation has confirmed the structural stability of the iturin A complex, whereby it exhibits less deviation from its structural conformation, less fluctuations, and a compact nature. The continued solvent exposure coupled with stable hydrogen bonding indicated that the ligand maintained its shape throughout the simulation. Through MD simulation and MMPBSA analysis, the overall binding energy of the system was determined to be −58.136±42.564 kJ/moL, indicating sustained ligand-protein binding throughout the simulation. The system’s total van der Waals energy was −240.564±25.594 kJ/moL. Furthermore, the net binding energy for the complex predicted by MMGBSA was −26.7955 kJ/moL, in contrast to the MMPBSA result of −22.9620 kJ/moL, as depicted in Figure 6f.
3.6. PCA and GFE Calculation
PCA and GFE analyses were conducted to explore structural dynamics. Figure 7a and b illustrate the principal component graph derived through the diagonalization of the covariance matrix of atomic coordinates. Correspondingly, Figures 7c and d depict the GFE values for the Apo and ligand-bound proteins, providing insights into the stability of their respective confirmations. In Figure 7 the PCA for the MD simulation data comprising 2000 frames is presented. In Figures 7a and b, each dot represents a frame, with the darkest blue denoting the initial frame and the brightest yellow representing the final frame (2000). The results revealed a notable clustering of dots, suggesting minimal variance in structural dynamics. Figure 7b displays a more extensive sampling than Figure 7a, potentially attributed to the confirmation of ligands and proteins. The corresponding GFE landscapes are shown in Figure 7c and d. As shown in Figure 7c, an Apo protein exhibits a strong cluster in the energy well (dark purple), signifying a stable conformation. Similarly, Figure 7d illustrates a distinct well for the ligand-bound protein, indicating stable confirmation. GFE values ranging from 0 to 1 kJ/moL provide valuable insights into the energetic landscape, indicating the stability and affinity of the protein-ligand complex. This narrow range suggests well-defined and favorable binding, supporting the robustness of the interaction in our study. In summary, both PCA and GFE analyses supported the notion of stable binding between the ligand and protein.
| | Figure 7: (a and b) Show the principal component graph derived through the diagonalization of the covariance matrix of the atomic coordinates. While (c and d) present the corresponding Gibbs free energy values for the Apo and ligand-bound protein, respectively. [Click here to view] |
3.7. Cytotoxic Effect of Recombinant Iturin A
The recorded cell viability percentages under the effective concentration range of iturin A (3.125–100 μg/mL) were analyzed, revealing a dose-dependent effect on breast cancer MCF-7 cells, as depicted in Figure 8. Notably, at a 100 μg/mL iturin A concentration, a remarkable cell viability of 13.2% was observed, surpassing the standard doxorubicin, which exhibited 23.69% cell viability at the same concentration. An IC50 value of 42.79 μg/mL further indicated 50% inhibition of cell proliferation by iturin A, with a discernible reduction in cell viability observed above 12.5 μg/mL, reinforcing its potential as an anticancer agent for breast cancer.
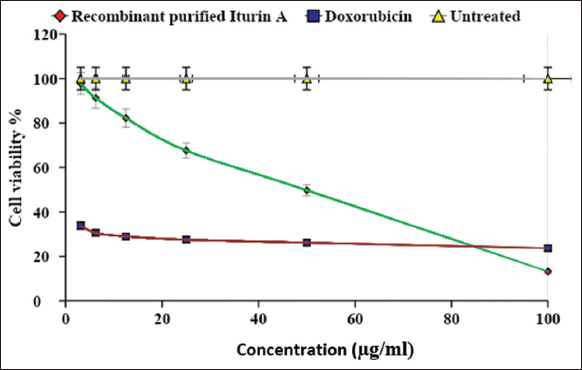 | Figure 8: Cytotoxicity assessment of recombinant iturin A against breast cancer cell line MCF-7. [Click here to view] |
Crucially, no significant inhibitory activity was detected in normal mammalian L929 cells within the defined concentration range of iturin A, maintaining 100% cell viability throughout the experiment. These findings highlight the selective anticancer potential of iturin A in breast cancer cells.
Doxorubicin, a standard chemotherapeutic drug, was used as the positive control in our experiments. While effective in suppressing breast cancer through apoptosis and necrosis, its use is hindered by undesirable side effects, such as cardiotoxicity. This study highlights the potential application of iturin A as a promising anticancer molecule with enhanced efficacy and reduced adverse effects, providing a foundation for future exploration and application in cancer treatment.
3.8. Apoptosis
Recombinant iturin was used to investigate apoptosis in MCF-7 cells, and the results showed a considerable increase in apoptosis. The IC50 value of 42.79 μg/mL was used as the treatment dose, which had a significant apoptotic impact. Flow cytometry analysis showed that the total apoptosis rate induced by recombinant iturin was 17.32%. The early and late apoptosis were observed to be 15.3% and 2.02%, respectively. In addition, 7.62 % was the percentage of dead cells, whereas the rest of the cells were alive, constituting a total population of 75.1%. In contrast, untreated MCF-7 cell lines showed no apoptosis, and the live cell percentage was 94.6%, while the remaining percentage was dead cells (Figure 9a and b).
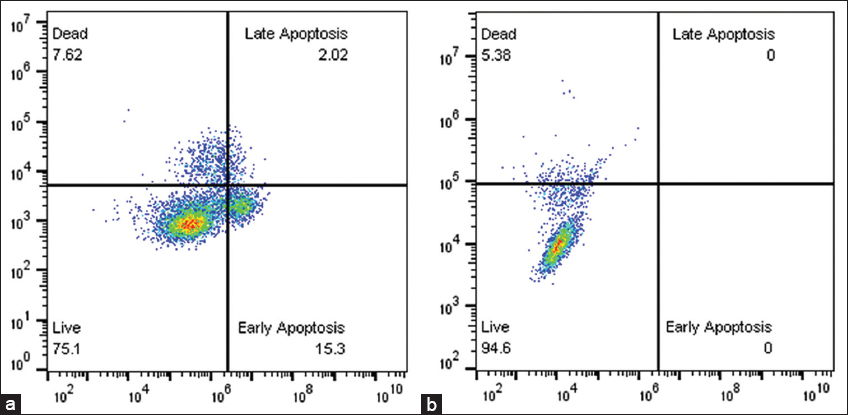 | Figure 9: (a) Induction of apoptosis in MCF-7 cells by recombinant iturin A. (b) Untreated MCF-7 cells. [Click here to view] |
4. DISCUSSION
Although various treatment strategies for breast cancer have been developed, the persistent increase in the relative incidence rate necessitates ongoing research. This study aimed to unravel the molecular underpinnings of breast cancer action, focusing on the PGR from Homo sapiens. By employing a combination of chemo-informatics and systems biology approaches, including target identification, target modeling, network pharmacology, gene set enrichment, and molecular docking, we unveiled the network-based effects of multi-component medications by analyzing drug-disease interactions in statistical databases. The prediction of the active component compositions and their targets facilitated by network analysis enables the identification of key drugs and disease targets. In particular, network pharmacology offers significant insights into its effects on PGR (4OAR). Despite the therapeutic potential of PGR in traditional medicine, few studies have focused on its unique activity against cancer cells. This in silico study compares the interaction of the PGR with a selective inhibitor and the beneficial molecule iturin A. The network pharmacology approach provides a robust framework for understanding the therapeutic potential of iturin A. Target identification and pathway enrichment analyses revealed its involvement in critical pathways implicated in breast cancer progression, including the HIF-1, VEGF, mTOR, and PI3K-Akt signaling pathways. These pathways are essential in regulating angiogenesis, cell survival, and proliferation, underscoring the compound’s multi-target potential. The estrogen signaling pathway with the PGR as a central node, emerged as a particularly significant mechanism of action for iturin A, especially in hormone-responsive breast cancer subtypes. Clusan et al. reported that luminal cells express PGRs [47]. The compound’s ability to interact with key proteins within this pathway highlights its potential to modulate hormone-regulated processes that drive cancer progression. According to Liang et al., through network pharmacology validation, the study reveals that ECH has certain effects on breast cancer by regulating PI3K/AKT/HIF-1α/VEGF signaling pathway, and ECH might be a promising natural compound to treat breast cancer for its multi-target and multi-pathway effects [48].
According to Sakle et al., for the chosen genes, GO and KEGG analyses identified different pathways along with diseases and illnesses. The direct involvement of bioactive compounds in the analysis of breast cancer was demonstrated in a GO enrichment study [49]. As per Hu et al., enrichment analysis using KEGG and GO was performed to investigate the biological pathways and pathways connected to the discovered genes [50]. The study also constructed protein-ligand interaction networks to evaluate the physical connectivity among different proteins, shedding light on the molecular mechanisms underlying cellular functions. Through network pharmacology, docking analysis, and simulation studies, we conducted a comprehensive scientific investigation of the pharmacological mechanism of PGR (4OAR) with iturin A in treating breast cancer.
Remarkably, our findings on the binding affinity and MD of bioactive substances, when compared to those of the validated inhibitor, are highly positive. The docking search identified the best conformation with the lowest docking energy. Iturin A exhibits a favorable binding energy of −5.5 kcal/moL when docked with PGR (4OAR). The study meticulously considered H-bond interactions, which indicate a stable connection between the ligand and the protein, by presenting these interactions through 2-D pictures and protein-ligand interaction depictions using Discovery Studio 2020 Client and Chimaera software. In addition, this study aligns with Rasul et al., suggesting that PGR (4OAR) can be a viable target for breast cancer treatment [51]. In line with Yaraguppi et al., docking was performed using AutoDock with specific grid coordinates (X, Y, and Z), and a docking score of −10.4 kcal/moL was reported for iturin A against a fungal pathogen protein [52]. A docking study was performed between the cholesterol margarate ligand and 4OAR protein, which showed a docking score of −6.9 kcal/moL [53]
MD computations utilize various methods to assess the stability of the protein-ligand complexes, including RMSD, RMSF, RG, H-Bonds, SASA, and MMPSA. The average RMSDs for the PGR protein and the iturin A complex protein over the simulation period of 0–200 ns were 0.28 nm and 0.18 nm, respectively, and a stable RMSD trend was observed. Zarezade et al. (2018) conducted molecular dynamic studies for 100 ns of a complex consisting of the PGR and compounds ZINC00936598, ZINC00869973, and ZINC01020370. All complexes experienced an RMSD of 0.100–0.415 nm throughout the course of the 100 ns MD simulation. This suggests convergence and a well-equilibrated trajectory suitable for the MD analyses. The results obtained from the study were highly relatable to the study carried out by Yaraguppi et al. 2022 which showed the stable configuration of iturin A in complex with the target MUR-A with a maximum of 0.6nm of RMSD, with the RG being at 2.2 nm showing its stability during the simulation duration of 200ns [37]. The results showed that iturin-A, when subjected to MD in a complex with the target proteins, remained stable during the simulation. In addition, MD and simulation studies with PGR protein were carried out by Zarezade et al. 2018 [54] clearly showed the stability of the protein in terms of RMSD, RMSF, RG, and H-bonds, which demonstrated that the lead compounds preserved the stability of ligands ZINC00936598, ZINC00869973, and ZINC01020370 when in complex with PGR during simulation in comparison with the standard drug positive control of Levonorgestrel for the duration of 100 ns. The root means square fluctuation of the app and complex form of the PGR backbone was computed throughout the course of the journey to examine the flexibility of the residues of this structure throughout the simulation of 200 ns [54]. The root mean square fluctuation of c-alpha atoms in the APO and LIG complexes was displayed over a time range of 0–200 ns where peaks were observed for relatively flexible regions in the protein. Greater flexibility is revealed by residues with high RMSF values, whereas less flexibility is revealed by residues with low RMSF values, which indicates limitations on residue mobility throughout the MD simulation [54]. RG values for PGR and iturin A complexes were plotted against the simulated timescale of 0–200 ns. In addition, SASA values and H-bond results of the iturin A complex were displayed over the 0–200 ns range. The stability of the Iturin A complex was confirmed by MD simulation parameters. The RMSD remained within 0.15–0.2 nm, indicating minimal structural deviations. RMSF values ranged from 0.1 to 0.25 nm, suggesting limited fluctuations in amino acid residues. The Radius of Gyration remained ≤1.9 nm, highlighting the compact nature of the complex. The SASA values were stable within 135–140 nm2, demonstrating consistent solvent exposure. In addition, the formation of 2–4 H-bonds ensured stable ligand-protein interactions, collectively confirming that the ligand maintained a relatively constant shape throughout the simulation.
The study also evaluated the binding free energy of the protein and the ligand through MD-based MMPBSA to calculate the electrostatic energy, polar solvation energy, and binding energy, yielding values of −135.500±87.220 kJ/moL, 349.422±98.522 kJ/moL, and −58.136±42.564 kJ/moL, respectively. Further residue decomposition revealed key energy contributions from ASP-745, ASP-697, ASP-781, ASP-746, GLU-695, GLU-723, ASP-704, GLU-911, GLU-907, GLU-833, LEU-726, GLU-904, ASP-709, and GLU-843. These comprehensive analyses lay the groundwork for the development of a specific chemical for further experimentation in animal studies. In the study carried out by Zarezade et al. 2018 [54], it was observed that the binding energy of PGR with different ligands remained in the range of −62 and −59 which is highly comparable with the binding energy obtained in the present study. In the present study, regions with interacting sites showed RMSF at lower levels. Regions exhibiting reduced RMSF on ligand binding enhance confidence in the accuracy of molecular docking and dynamics results. According to Rasul et al. 2022, for RMSD, a value of <2 A is considered an accurate estimation for ligand-protein computational confirmation. Along with RMSD, the RMSF of the complex in the study showed steady results and plummeted less, with the largest RMSF value of 4.5 A [55].
This study aligns with existing literature reporting the anticancer potential of iturin A against hepatocellular carcinoma (HepG2 cells), suggesting mechanisms involving reactive oxygen species, apoptosis induction, and cell cycle disruption. In addition, studies on iturin A from Bacillus megaterium have highlighted its ability to suppress VEGF, inhibit MMP-2/9 expression, and interfere with the MD-2/TLR4 signaling pathway, as validated through in silico docking approaches [56]. In the present study, we investigated the anticancer activity of recombinant iturin against the MCF-7 cell line using an MTT assay. Our findings indicated a concentration-dependent decrease in anticancer activity, as cell viability decreased with increasing concentrations. Microscopic morphological studies also supported these findings, showing that the treated cells became morphologically distinct from their untreated counterparts and were shrunken and apoptotic. Recombinant iturin had an IC50 of 42.79 μg/mL. Iturin is a lipopeptide derived from Bacillus species that was discovered to possess anticancer activity. In breast cancer cell lines, such as MCF-7, iturin inhibits the cell remarkably, and it impacts the viability of cells as demonstrated by the MTT assay [11]. Since iturin possesses various excellent properties, such as antibacterial, antifungal, antiviral, anticancer, and anti-obesity activities, the pharmaceutical industry is quite aware of its usability. Among lipopeptides obtained from Bacillus species, iturin has been found to possess anticancer activity [57]. Apoptosis is a strictly controlled process, sometimes called “programmed cell death,” in which cells can die in an orderly fashion. It is essential for maintaining tissue homeostasis, eliminating diseased or toxic cells, and directing the growth of multicellular organisms [58]. In Figures 9a and b, the upper left part shows the dead cells, the lower left part indicates live cells, the upper right part indicates late apoptosis, and the lower right part indicates early apoptosis. When comparing treated and untreated cells, there was a significant decrease in the percentage of live cells from 94.6 to 75.1; on the other hand, there was an increase in the percentage of dead cells, late apoptosis, and early apoptosis. According to a study by Dan et al., in molecular defense against cancer, the lipopeptide iturin A may regulate the pathways leading to apoptosis within tumor cells by inducing DNA damage and ROS generation. Regarding the anticancer molecular mechanism, DNA damage and generation of ROS are related to the apoptotic pathways of tumor cells in two ways; further, iturin also triggers autophagy and pyroptosis through metabolic disruption that leads to programmed cell death of the cell [59]. Zhao et al. reported that iturin A treated HEPG2 cell line showed apoptosis with live cells of 75.13%, dead cells of 6.28%, and early apoptosis was 18.59% and there was no late apoptosis during this study [21].
Breast cancer MCF-7 cell lines have been widely employed in anticancer studies, and other researchers have explored the cytotoxic effects of diverse compounds, such as keratin hydrolysates and nanoparticles derived from hydrolysates [60]. Compounds from the roots of Withania somnifera have also demonstrated anticancer properties by targeting KAT6A, a critical player in key cellular pathways [61].
Limited research has been conducted on the interaction between PGR and iturin A for the treatment of breast cancer. The role of PR in breast cancer is commonly linked to hormonal therapy, and the exploration of its application in this context represents an emerging area of investigation [62]. PGR, a nuclear receptor activated by progesterone, contributes to the regulation of diverse biological processes, including those integral to the menstrual cycle and pregnancy [63]. Assessment of PGRs in breast cancer serves as a crucial component of hormone receptor status, with hormone receptor-positive breast cancers, including those expressing PGR, which are potentially responsive to hormone therapies [64]. Iturin A, recognized for its antifungal properties, holds promise as a potential anticancer agent, particularly in breast cancer. However, the specific interaction between iturin A and PGRs in the context of breast cancer treatment has not been extensively studied and documented. Further research is imperative to unravel the intricate details and implications of this interaction.
Our study constructed proteins-ligand-pathways interaction networks to address this gap, offering insights into the physical connectivity among different proteins. This approach sheds light on the molecular mechanisms governing cellular functions. By employing network pharmacology, docking analysis, and simulation studies, we undertook a comprehensive scientific investigation into the pharmacological mechanisms of iturin A in breast cancer treatment. Further studies such as MTT assay and apoptosis were carried out to check the activity of iturin A as a potent anti-cancer drug for breast cancer, and the results revealed that iturin A can be used as a drug for breast cancer treatment. Iturin A exerts direct effects on breast cancer cells, its potential modulation of hormone receptor activity, and its interactions with other pathways pivotal to breast cancer progression. Through ongoing research, we aim to enhance our understanding of its role in breast cancer treatment and its broader therapeutic implications.
5. CONCLUSION
The integration of network analysis, structure-based drug design, and simulation analysis has been employed with precision in recent research. The results derived from this study rest on the application of accurate and reliable software, substantiating their utility. The initial utilization of network analysis, molecular docking with docking score of −5.5 kcal/moL, MD, and simulation studies proved to be a cost-effective and efficient alternative, mitigating the need for extensive in vitro experiments. The novel compound exhibiting promising outcomes warrants further exploration through MD simulations to capture real-time behavioral dynamics. Subsequent in vitro validation was performed to not only confirm the computational findings but also unveil a new frontier in drug design, particularly for effective breast cancer therapeutics. The in silico approach employed in this study sheds light on the various potential mechanisms of PGR as a prospective target for breast cancer. This study not only establishes a theoretical framework for comprehending the underlying molecular simulation mechanisms but also lays the foundation for the identification of potential targets and drugs, addressing the needs of breast cancer patients. It is important to note that the conclusions drawn in this study were based on computational simulations and in vitro studies. Furthermore, cell proliferation and in vivo investigations can be performed to further confirm the anticancer activity of iturin A.
6. AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
7. FUNDING
There is no funding to report.
8. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
9. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
10. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
11. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
12. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Enayatrad M, Amoori N, Salehiniya H. Epidemiology and trends in breast cancer mortality in iran. Iran J Public Health 2015;44:430-1.
2. Sant M, Allemani C, Capocaccia R, Hakulinen T, Aareleid T, Coebergh JW, et al. Stage at diagnosis is a key explanation of differences in breast cancer survival across Europe. Int J Cancer 2003;106:416-22.[CrossRef]
3. Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2019;2:141-60.[CrossRef]
4. Dehelean CA, Marcovici I, Soica C, Mioc M, Coricovac D, Iurciuc S, et al. Plant-derived anticancer compounds as new perspectives in drug discovery and alternative therapy. Molecules 2021;26:1109.[CrossRef]
5. Yang JY, Ha SA, Yang YS, Kim JW. -Glycoprotein ABCB5 and YB-1 expression plays a role in increased heterogeneity of breast cancer cells:Correlations with cell fusion and doxorubicin resistance. BMC Cancer 2010;10:388.[CrossRef]
6. Tsai SY, Carlstedt-Duke J, Weigel NL, Dahlman K, Gustafsson JA, Tsai MJ, et al. Molecular interactions of steroid hormone receptor with its enhancer element:Evidence for receptor dimer formation. Cell 1988;55:361-9.[CrossRef]
7. Tseng L, Tang M, Wang Z, Mazella J. Progesterone receptor (hPR) upregulates the fibronectin promoter activity in human decidual fibroblasts. DNA Cell Biol 2003;22:633-40.[CrossRef]
8. Richer JK, Lange CA, Manning NG, Owen G, Powell R, Horwitz KB. Convergence of progesterone with growth factor and cytokine signaling in breast cancer. Progesterone receptors regulate signal transducers and activators of transcription expression and activity. J Biol Chem 1998;273:31317-26.[CrossRef]
9. Gellersen B, Fernandes MS, Brosens JJ. Non-genomic progesterone actions in female reproduction. Hum Reprod Update 2009;15:119-38.[CrossRef]
10. Cao XH, Wang AH, Wang CL, Mao DZ, Lu MF, Cui YQ, et al. Surfactin induces apoptosis in human breast cancer MCF-7 cells through a ROS/JNK-mediated mitochondrial/caspase pathway. Chem Biol Interact 2010;183:357-62.[CrossRef]
11. Dey G, Bharti R, Dhanarajan G, Das S, Dey KK, Kumar BN, et al. Marine lipopeptide Iturin A inhibits Akt mediated GSK3b and FoxO3a signaling and triggers apoptosis in breast cancer. Sci Rep 2015;5:10316.[CrossRef]
12. Jacobsen BM, Schittone SA, Richer JK, Horwitz KB. Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer:PR isoform-specific gene regulation and tumor biology. Mol Endocrinol 2005;19:574-87.[CrossRef]
13. Migliaccio A, Piccolo D, Castoria G, Di Domenico M, Bilancio A, Lombardi M, et al. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J 1998;17:2008-18.[CrossRef]
14. Taraborrelli S. Physiology, production and action of progesterone. Acta Obstet Gynecol Scand 2015;94(Suppl 161):8-16.[CrossRef]
15. Brisken C, Park S, Vass T, Lydon JP, O'Malley BW, Weinberg RA. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci U S A 1998;95:5076-81.[CrossRef]
16. Brisken C. Progesterone signalling in breast cancer:A neglected hormone coming into the limelight. Nat Rev Cancer 2013;13:385-96.[CrossRef]
17. Zhao H, Shao D, Jiang C, Shi J, Li Q, Huang Q, et al. Biological activity of lipopeptides from Bacillus. Appl Microbiol Biotechnol 2017;101:5951-60.[CrossRef]
18. Kim SY, Kim JY, Kim SH, Bae HJ, Yi H, Yoon SH, et al. Surfactin from Bacillus subtilis displays anti-proliferative effect via apoptosis induction, cell cycle arrest and survival signaling suppression. FEBS Lett 2007;581:865-71.[CrossRef]
19. Vo TT, Wee Y, Cheng HC, Wu CZ, Chen YL, Tuan VP, et al. Surfactin induces autophagy, apoptosis, and cell cycle arrest in human oral squamous cell carcinoma. Oral Dis 2023;29:528-41.[CrossRef]
20. Hajare SN, Subramanian M, Gautam S, Sharma A. Induction of apoptosis in human cancer cells by a Bacillus lipopeptide bacillomycin D. Biochimie 2013;95:1722-31.[CrossRef]
21. Zhao H, Yan L, Guo L, Sun H, Huang Q, Shao D, et al. Effects of Bacillus subtilis iturin A on HepG2 cells in vitro and in vivo. AMB Express 2021;11:67.[CrossRef]
22. Hopkins AL. Network pharmacology:The next paradigm in drug discovery. Nat Chem Biol 2008;4:682-90.[CrossRef]
23. Zhang R, Zhu X, Bai H, Ning K. Network pharmacology databases for traditional chinese medicine:Review and assessment. Front Pharmacol 2019;10:123.[CrossRef]
24. Jiashuo WU, Fangqing Z, Zhuangzhuang LI, Weiyi J, Yue S. Integration strategy of network pharmacology in traditional Chinese medicine:A narrative review. J Tradit Chin Med 2022;42:479-86.
25. Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking:A powerful approach for structure-based drug discovery. Curr Comput Aided-Drug Des 2011;7:146-57.[CrossRef]
26. Adam Hospital, Goñi JR, Orozco M, GelpíJL. Molecular dynamics simulations:Advances and applications. Adv Appl Bioinform Chem 2015;8:37-47.[CrossRef]
27. Mithilesh S, Raghunandan D, Suresh PK. In-silico identification of natural compounds from traditional medicine as potential drug leads against SARS-CoV-2 through virtual screening. Proc Natl Acad Sci India Sect B Biol Sci 2022;92:81-7.[CrossRef]
28. Yan L, Zhang Z, Liu Y, Ren S, Zhu Z, Wei L, et al. Anticancer activity of erianin:Cancer-specific target prediction based on network pharmacology. Front Mol Biosci 2022;9:862932.[CrossRef]
29. Luo Y, Fu Y, Tan T, Hu J, Li F, Liao Z, et al. Screening of lncRNA-miRNA-mRNA coexpression regulatory networks involved in acute traumatic coagulation dysfunction based on CTD, GeneCards, and PharmGKB databases. Oxid Med Cell Longev 2022;2022:7280312.[CrossRef]
30. Jia A, Xu L, Wang Y. Venn diagrams in bioinformatics. Brief Bioinform 2021;22:bbab108.[CrossRef]
31. Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, et al. The STRING database in 2023:Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 2023;51:D638-46.[CrossRef]
32. Huang F, Fu M, Li J, Chen L, Feng K, Huang T, et al. Analysis and prediction of protein stability based on interaction network, gene ontology, and KEGG pathway enrichment scores. Biochim Biophys Acta Proteins Proteom 2023;1871:140889.[CrossRef]
33. Piñero J, Saüch J, Sanz F, Furlong LI. The DisGeNET cytoscape app:Exploring and visualizing disease genomics data. Comput Struct Biotechnol J 2021;19:2960-7.[CrossRef]
34. Acharya R, Chacko S, Bose P, Lapenna A, Pattanayak SP. Structure based multitargeted molecular docking analysis of selected furanocoumarins against breast cancer. Sci Rep 2019;9:15743.[CrossRef]
35. Ibrahim MA, Badr EA, Abdelrahman AH, Almansour NM, Mekhemer GA, Shawky AM, et al. In silico targeting human multidrug transporter ABCG2 in breast cancer:Database screening, molecular docking, and molecular dynamics study. Mol Inform 2022;41:2060039.[CrossRef]
36. Patidar M, Yadav N, Dalai SK. Designing of suitable linkers for the chimeric proteins to achieve the desired flexibility and extended conformation. Int J Comput Biol Drug Des 2018;11:236.[CrossRef]
37. Yaraguppi DA, Bagewadi ZK, Deshpande SH, Chandramohan V. In silico study on the inhibition of UDP-N-acetylglucosamine 1-carboxy vinyl transferase from Salmonella typhimurium by the lipopeptide produced from Bacillus aryabhattai. Int J Pept Res Ther 2022;28:80.[CrossRef]
38. Amadei A, Linssen AB, Berendsen HJ. Essential dynamics of proteins. Proteins 1993;17:412-25.[CrossRef]
39. Rajendran V, Sethumadhavan R. Drug resistance mechanism of PncA in Mycobacterium tuberculosis. J Biomol Struct Dyn 2014;32:209-21.[CrossRef]
40. Khan MT, Ali S, Zeb MT, Kaushik AC, Malik SI, Wei DQ. Gibbs free energy calculation of mutation in PncA and RpsA associated with pyrazinamide resistance. Front Mol Biosci 2020;7:52.[CrossRef]
41. Sun H, Li Y, Shen M, Tian S, Xu L, PanP, et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys Chem Chem Phys 2014;16:22035-45.[CrossRef]
42. Martis EAF, Coutinho EC. Free energy-based methods to understand drug resistance mutations. In:Structural Bioinformatics:Applications in Preclinical Drug Discovery Process. Cham:Springer;2019. 1-24.[CrossRef]
43. Yaraguppi DA, Bagewadi ZK, Mahanta N, Singh SP, Khan TM, Deshpande SH, et al. Gene expression and characterization of Iturin a lipopeptide biosurfactant from Bacillus aryabhattai for enhanced oil recovery. Gels 2022;8:403.[CrossRef]
44. Shettar SS, Bagewadi ZK, Yaraguppi DA, Das S, Mahanta N, Singh SP, et al. Gene expression and molecular characterization of recombinant subtilisin from Bacillus subtilis with antibacterial, antioxidant and anticancer properties. Int J Biol Macromol 2023;249:125960.[CrossRef]
45. Bagewadi ZK, Muddapur UM, Madiwal SS, Mulla SI, Khan A. Biochemical and enzyme inhibitory attributes of methanolic leaf extract of Datura inoxia Mill. Environ Sustain 2019;2:75-87.[CrossRef]
46. Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, et al. PubChem substance and compound databases. Nucleic Acids Res 2016;44:D1202-13.[CrossRef]
47. Clusan L, Ferrière F, Flouriot G, Pakdel F. A basic review on estrogen receptor signaling pathways in breast cancer. Int J Mol Sci 2023;24:6834.[CrossRef]
48. Liang H, Yin G, Shi G, Liu Z, Liu X, Li J. Echinacoside regulates PI3K/AKT/HIF-1a/VEGF cross signaling axis in proliferation and apoptosis of breast cancer. Anal Biochem 2024;684:115360.[CrossRef]
49. Sakle NS, More SA, Mokale SN. A network pharmacology-based approach to explore potential targets of Caesalpinia pulcherrima:An updated prototype in drug discovery. Sci Rep 2020;10:17217.[CrossRef]
50. Hu H, Wang H, Yang X, Li Z, Zhan W, Zhu H, et al. Network pharmacology analysis reveals potential targets and mechanisms of proton pump inhibitors in breast cancer with diabetes. Sci Rep 2023;13:7623.[CrossRef]
51. Rasul MF, Hussen BM, Salihi A, Ismael BS, Jalal PJ, Zanichelli A, et al. Strategies to overcome the main challenges of the use of CRISPR/Cas9 as a replacement for cancer therapy. Mol Cancer 2022;21:64.[CrossRef]
52. Yaraguppi DA, Deshpande SH, Bagewadi ZK, Kumar S, MuddapurUM.Genome analysis of Bacillus aryabhattai to identify biosynthetic gene clusters and in silico methods to elucidate its antimicrobial nature. Int J Pept Res Ther 2021;27:1331-42.[CrossRef]
53. Raju L, Lipin R, Eswaran R. Identification, ADMET evaluation and molecular docking analysis of phytosterols from banaba (Lagerstroemia speciosa (L.)Pers) seed extract against breast cancer. In Silico Pharmacol 2021;9:43.[CrossRef]
54. Zarezade V, Abolghasemi M, Rahim F, Veisi A, Behbahani M. In silico assessment of new progesterone receptor inhibitors using molecular dynamics:A new insight into breast cancer treatment. J Mol Model 2018;24:337.[CrossRef]
55. Rasul HO, Aziz BK, Ghafour DD, Kivrak A. Correction to:In silico molecular docking and dynamic simulation of eugenol compounds against breast cancer. J Mol Model 2022;28:78.[CrossRef]
56. Dey G, Bharti R, Ojha PK, Pal I, Rajesh Y, Banerjee I, et al. Therapeutic implication of 'Iturin A'for targeting MD-2/TLR4 complex to overcome angiogenesis and invasion. Cell Signal 2017;35:24-36.[CrossRef]
57. Yaraguppi DA, Bagewadi ZK, Patil NR, Mantri N. Iturin:A promising cyclic lipopeptide with diverse applications. Biomolecules 2023;13:1515.[CrossRef]
58. Elmore S. Apoptosis:A review of programmed cell death. Toxicol Pathol 2007;35:495-516.[CrossRef]
59. Dan AK, Manna A, Ghosh S, Sikdar S,Sahu R, Parhi PK, et al. Molecular mechanisms of the lipopeptides from Bacillus subtilis in the apoptosis of cancer cells - A review on its current status in different cancer cell lines. Adv Cancer Biol Metastasis 2021;3:100019.[CrossRef]
60. Ferroni C, Varchi G. Keratin-based nanoparticles as drug delivery carriers. Appl Sci 2021;11:9417.[CrossRef]
61. Deshpande SH, Muhsinah AB, Bagewadi ZK, Ankad GM, Mahnashi MH, Yaraguppi DA, et al. In silico study on the interactions, molecular docking, dynamics and simulation of potential compounds from Withania somnifera (L.) root against cancer by targeting KAT6A. Molecules 2023;28:1117.[CrossRef]
62. Horwitz KB, Sartorius CA. 90 years of progesterone:Progesterone and progesterone receptors in breast cancer:Past, present, future. J Mol Endocrinol 2020;65:T49-63.[CrossRef]
63. Dinh DT, Breen J, Nicol B, Foot NJ, Bersten DC, Emery A, et al. Progesterone receptor mediates ovulatory transcription through RUNX transcription factor interactions and chromatin remodelling. Nucleic Acids Res 2023;51:5981-96.[CrossRef]
64. Li Z, Wei H, Li S, Wu P, Mao X. The role of progesterone receptors in breast cancer. Drug Des Devel Ther 2022;16:305-14.[CrossRef]