
1. INTRODUCTION
Cytochrome P450 (CYPs) are heme-containing monooxygenases that contribute to vital life processes with their compelling role in biotransformation or metabolism of various compounds of both xenobiotic and endogenous origin, apart from their involvement in the biosynthesis of fatty acids, sterols, vitamins, eicosanoids, etc. [1,2]. In humans, these hemoproteins are present primarily at high concentrations in the liver along with their differential expressions in various tissues such as skin, lung, kidney, heart, gastrointestinal tract, nasal mucosa, and brain. The CYP enzyme subcellular localization is typically membrane of endoplasmatic reticulum and mitochondria [3]. These enzymes execute a significant act in Phase I drug metabolism, oxidizing 70–80% of pharmaceutical drug metabolism reactions in humans [3,4].
Among the 57 CYP isoforms, CYP3A4 is a considerable drug-metabolizing CYP, associated with the biotransformation of 30% drugs consumed orally, succeeded by the other isoforms CYP1A2, CYP2D6, and CYP2C9 [5,6]. CYP3A4 is the most abundant liver and intestinal CYP which has very broad substrate specificity and can metabolize molecules widely differing in size and chemical structure through alkyl carbon and aromatic ring hydroxylation, O- and N-dealkylation, and epoxidation [7]. In addition, CYP3A4 can concurrently adapt numerous molecules at the active site which might impel to coordinated binding and also atypical (non-Michaelis–Menten) kinetic attitude [7]. Its disparate specificity and collective binding of substrates are currently undergoing undesirable drug-drug intercommunications, along with lethal side effects. In the case of new prospective medications, monitoring drug-drug interactions is critical [8].
Drug responsiveness, effectiveness, and adverse-effect rates are influenced by interindividual heterogeneity in CYP3A4 activity and expression. CYP3A4 genetic variations alter drug responses, in addition to the other factors that contribute to this variability, such as CYP3A4 inducers and inhibitors, as well as food drug and drug-drug interactions [9,10]. Deletions, insertions, copy number variants, and single nucleotide polymorphisms (SNPs) in the coding and non-coding sections of the genes are examples of variant alleles that can affect CYP450 expression levels and protein function [11]. For CYP3A4 isoforms, over 100 non-synonymous single amino acid changes have been documented [12]. Enzymatic activity may be abolished, reduced, altered, or augmented as a result of mutations in the CYP3A4 gene. Exonic mutations have been shown to change enzyme activity in a few clinical investigations with specific substrates, and altered activity due to CYP3A4 mutations has already been described in the literature [13-16]. The use of probe medicines to assess CYP3A4 activity is critical for determining the influence of genetic variation on inter-individual variability in CYP3A4 activity. Hence, in this study, Imipramine, Midazolam, Nifedipine, and Quinidine compounds were chosen to analyze the impact of mutations in the CYP3A4 activity and ligand binding. In this study, CYP3A4 mutants were created and analyzed in silico for their increased stability and efficiency so that they can have a profound response on binding to the ligand molecules. Hence, multiple sequence-based alignments to know the single amino acid substitutions and subsequently, mutational assessment using various tools to verify the stability and favorability of the identified mutations were carried out. Molecular modeling and docking approaches were implemented with chosen ligands to know their interactions with the wild and mutant protein models.
2. MATERIALS AND METHODS
2.1. Data Retrieval of the Target Protein
The three-dimensional (3D) structure and sequence of target protein human CYPs 3A4 were retrieved from the protein data bank (PDB) and Uniprot database, respectively. The structural data of human microsomal P450 3A4 were taken from PDB with corresponding PDB ID 1TQN and solved by X-ray diffraction at 2.05 Å [17]. The crystal structure of human microsomal P450 3A4 is shown in Figure 1. The protein reference sequence was retrieved in FASTA format from Uniprot (ID: P08684). For the identification of similar sequences, the homology search of query protein was performed using the BLASTP algorithm against UniProtKB/Swiss-Prot, with the default parameters. Four sequences have been selected based on their similarity and retrieved their sequences in Fasta format.
 | Figure 1: 3D structures of the protein structure (1TQN). The cofactor heme is depicted in sticks. [Click here to view] |
2.2. Multiple Sequence Alignment (MSA)
From the results of BLAST against the Swiss-prot database, four sequences have been selected with varying sequence percent identity in the range of 100–85% with the sequence of human CYP3A4. Multiply aligning the sequences gives insight into conserved and non-conserved amino acid sites. The Clustal Omega program [18] was used for MSA with default settings between the selected sequences. The retrieved sequences are submitted to Clustal Omega which determines the better match for the preferred sequences and calculates the similarities identities, and differences can be observed. Based on these results, single-site mutations were created in the conserved regions of the target protein. Multiple sequence analysis identified the consensus and conserved regions between the selected sequences. All the sequences including target protein human CYP3A4 showed conserved ‘*’ (asterisk) and non-conserved ‘:’ (colon) amino acids positions Among the substitutions, only ‘:’ positions which resulted by substitution with similar physiochemical properties of amino acids were considered for further study and ‘.’ (period) that represents change with weakly similar amino acid residues that were left out. Based on the results of MSA in Figure 2, residues that shown different substitutions in the target sequence and also occurring with different substitutions in other sequences taken for the present study. From the screening results, 40 single-site mutations were taken for analysis. H54R, H65Y, L129I, M256I, E262K, H28R, S29T, F46L, Y53F, K55Q, F57Y, V71M, V81M, M89I, E122Q, E124D, Q151K, D154E, V170I, L172M, V175I, D214N, L229I, V253I, R255Q, D263E, Q265H, S286T, V296M, L331Q, E333Q, M353L, L366I, K422N, K424N, S437T, M450N, L456I, I457V, and l479F.
 | Figure 2: Multiple sequence alignment of the taken sequences and CYP3A4 (P08684). [Click here to view] |
2.3. Stability Prediction
Mutations can ensure alterations in the stability of the protein structure. Protein stability is the necessary aspect that affects the activity, function, and regulation of biological molecules. A key indicator of protein stability is the unfolding free energy of the protein. Analyzing the free energy change on mutation, the impact of the mutation on protein stability could be precisely estimated. For calculating protein stability changes using thermodynamic cycle (?GF wt to mt), subtracting the mutant protein free energy change from wild protein free energy change expressed in Kcal/mol (DDG or ??G) = ?G mutant – DG wild type). For a given protein mutation, the DDG value of above zero predicts high stability and a score below zero predicts low stability. To check the stability of mutations in terms of energy, two stability prediction tools Site-Directed Mutation (SDM) [19] and Cologne University protein Stability Analysis Tool (CUPSAT) [20] have been used in the present study. Further, the SDM stabilizing mutations are analyzed with CUPSAT, which predicts the stabilized and favorable mutations.
SDM examines the assortment of amino acid substitutions happening at fundamental circumstances that are tolerated within the family of known 3D structures of homologous proteins and displays them as unconstructive substitution tables. As a quantifiable measure, these tables have been used to predict the protein structure on an alteration in amino acid. This server depends on the examination of ??G (in kcal/mol) which gives its values of stabilizing mutations (??G > 2.5 kcal/mol) and destabilizing changes (??G < −2.5 kcal/mol). SDM discriminate the competency of amino acid residues among the wild and mutant proteins.
CUPSAT determines the stability based on torsional angle potential, solvent accessibility, and difference in wild and mutant proteins free energy on unfolding. It requires a PDB structure and the position of the substituted residue for finding favorable and stabilizing mutations. The output encompasses data about mutation sites, their structural features, and includes information about the change in the stability of each residue with the rest of 19 amino acids.
In the present study, the impact of the 40 single site mutations on CYPs 3A4 (1TQN) protein stability changes was examined, to evaluate their effect on protein folding. The unfolding DDG was calculated. The protein sequence and/or protein structure of CYPs 3A4 (1TQN) with mutational position and amino acid residues to be mutated were used to test the predictive value of stability prediction programs using SDM and CUPSAT online tools.
For the final predictive results of the two tools, a restricted criterion was set. When the results of the above tools were integrated, the mutation that was predicted to be “stabilizing” by the two in silico tools would be considered and was selected for further analysis.
2.4. Analyzing Protein Evolutionary Conservation
To establish the amino acids evolutionary conservation in the CYP3A4 protein sequence, the ConSurf tool [21] was implemented that accomplishes its function by examining the phylogenetic relationships among similar sequences. Initially, it distinguishes the conserved positions with the aid of MSA, then using empirical Bayesian inference, the evolutionary conservation rate was calculated and presents the profiles of evolutionary conservation of sequence or the structure of the protein. According to the Bayesian approach, three different categories of conservation scores are applied, 7–9 score indicates conservedness, 5–6 score indicates intermediate, and 1–4 score implies variable. Inclusive of conservation profile, the regions of proteins such as buried and exposed are also predicted. Along with these results, the tool also provides the functional/structural impact of the amino acid across the protein.
2.5. Modeling and of Mutant Structures
The protein 3D structure is important to analyze the functionality, particularly when trying to interpret the effect of mutations on its overall structure and function. To make the mutated models of the human CYP3A4 for correspondent amino acid replacements, the “Build Mutant” protocol integrated with the DS modeling settings was implemented to substitute the wild type amino acid with a new amino acid. The modeling of the mutated structures of human CYP3A4 was built using the wild type available 3D structure (1TQN) as a reference. The protocol “Built Mutant” adopts the modeler program to mutate residues to particularized residues and ameliorates the mutated and neighboring residues conformation. Further to improve the quality of the predicted models, energy minimization studies were carried out.
2.6. Wild and Mutant Proteins Preparation and Validation
The wild and mutant proteins are imported into Accelrys Discovery Studio 2.1 and using the protocol’s clean protein, the structures are prepared individually. Before processing for preparation, removal of the heteroatoms, water molecules, and inbuilt ligands is done. Addition of hydrogen atoms to the protein structures interrelated to pH = 7.4. Then the protocol accomplishes protein structure processing that refines their bond orders, standardizing names of the atoms, inserting missing atoms in incomplete residues, modeling missing loop regions, deleting alternate conformations, and protonating titratable residues. Finally, all atom restrained protein structure energy minimization is performed applying CHARMm force field with different algorithms such as steepest descent preceded by conjugant gradient with maximum steps of 1000 for each algorithm, respectively, until the convergence gradient is satisfied with a root to mean square deviation (RMSD) tolerance of 0.01 Å.
The quality of energy-minimized protein structures was determined with the use of validation tools such as Procheck, ProSA, and RMSD of the mutant models concerning the wild CYP3A4 was calculated using SPDBV.
2.7. Active Site Identification
After energy minimization, with the use of the DS, Define and Edit Binding Site tool, the active site of the protein was selected on the base of the inbuilt active site pocket conformation and with a radius of 10 Å, respectively, an active site sphere was described.
2.8. Ligand Preparation
The compounds chosen in the present study were Imipramine, Midazolam, Nifedipine, and Quinidine. The chemical structures of the compounds are sketched using ACD/ChemSketch (12.0) Software and saved in mol2 format. The saved compounds are later imported into DS and their 2D structures were converted to 3D representation by using the catalyst algorithm of DS. Prepare ligands module of DS was used for ligand preparation which corrects for hydrogen bonds (HB) addition, bond lengths, bond angles, isomer, and tautomer generation and filters the ligands by removing the duplicate structures. Further followed by minimization and optimization in CHARMm force field with the smart minimizer algorithm which follows by the conjugant gradient algorithm, till it was satisfied with the convergence gradient of 0.001 kcal/mol for attaining the low energy conformational structures. The three-dimensional structures of the compounds are depicted in Figure 3.
 | Figure 3: 3D structures of the chosen ligands. [Click here to view] |
2.9. Molecular Docking
A molecular docking approach is implemented to depict the binding affinities along with their interaction modes of chosen ligands with the CYP3A4 wild and mutant model proteins with the aid of the DS LibDock module. To know the catalytic activeness of a ligand, the ligand-protein binding patterns and interactions are very important.
The minimized wild and mutant protein models, as well as ligands along with the binding site coordinates or the X, Y, and Z points of the binding site residues within 10 Å, are submitted individually to the LibDock setup. All the other parameters of docking and consequence scoring functions applied were accomplished at their default environment. The active site features are indicated as “HotSpots” and that is persistent in a grid settled innermost the active site. This algorithm aligned the ligand configuration to hotspots (polar and a polar) of receptor interaction sites and retains the best protein-ligand complex pose at the end of the docking process.
All the retained complex poses of the protein and compounds are ranked according to the LibDock scoring function for binding affinity calculations that are studied to determine the potentiality of the molecules docked. Libdock score is used for estimating the binding affinity of each pose and binding pose with the topmost LibDock score and lowest binding energy are adopted as the best pose for each ligand and their binding interactions are analyzed. The interaction poses of the best complex pose are studied in terms of intermolecular HBs and close contacts.
3. RESULTS AND DISCUSSION
To know the sequence and structure-based stability of mutation, the structure, and its reference sequences were retrieved from the PDB (1TQN) and UniProt (UniProt ID: P08684) database respectively. Although RCSB reported that, 1TQN (the crystal structure of human microsomal CYP3A4) consists of the amino acid residues 28-499 and the PO8684 (CYP3A4 sequence) consists of 503 amino acid residues.
3.1. Stability Prediction
Site-Directed Mutator (SDM server) computes the comprised study of wild-type and mutated secondary structure evaluation, solvent accessibility, depth (Å) of the protein, and ??G value which predicts the stability or disability of protein. SDM predicts ??G scores which revealed that 17 out of 40 mutants have shown increased stability of protein and all the remaining mutated residues have shown decreased stability of protein as shown in Table 1. The resulting outcome mostly residues have a negative value which predicted that may be a mutation on these residues of a point caused protein malfunction.
Table 1: The predicted structure stability ??G and topological properties in between wild type and mutant (secondary structure, solvent accessibility and depth) using SDM server.
| Mutation | Wild type | Mutant type | Predicted DDG | Outcome | ||||
|---|---|---|---|---|---|---|---|---|
| Secondary structure | Relative solvent accessibility | Residue depth (Å) | Secondary structure | Relative solvent accessibility | Residue depth (Å) | |||
| H54R | H | 80.5 | 3.4 | H | 86.7 | 3.3 | 0.15 | Increased stability |
| H65Y | H | 18.7 | 4.3 | H | 24.8 | 4.4 | 0.91 | Increased stability |
| L129I | H | 0.0 | 7.3 | H | 0.0 | 7.6 | −0.51 | Reduced stability |
| M256I | H | 3.8 | 6.6 | H | 4.5 | 6.2 | −0.29 | Reduced stability |
| E262K | a | 62.5 | 3.6 | H | 81.4 | 3.3 | −0.82 | Reduced stability |
| H28R | b | 65.3 | 3.3 | g | 18.0 | 4.9 | 0.0 | Increased stability |
| S29T | a | 63.1 | 3.6 | a | 41.7 | 3.8 | 0.05 | Increased stability |
| F46L | g | 90.0 | 3.4 | g | 80.8 | 3.4 | 0.38 | Increased stability |
| Y53F | H | 4.3 | 7.0 | H | 3.3 | 7.0 | −0.07 | Reduced stability |
| K55Q | H | 57.1 | 3.6 | H | 44.8 | 3.9 | 0.44 | Increased stability |
| F57Y | H | 12.0 | 6.0 | H | 12.9 | 6.2 | 0.12 | Increased stability |
| V71M | E | 6.7 | 4.5 | E | 8.5 | 4.8 | −2.25 | Reduced stability |
| V81M | E | 0.0 | 8.8 | E | 0.0 | 8.4 | −1.75 | Reduced stability |
| M89I | H | 4.0 | 5.7 | H | 0.8 | 6.0 | −0.36 | Reduced stability |
| E122Q | b | 48.4 | 3.6 | b | 60.0 | 3.8 | 0.11 | Increased stability |
| E124D | H | 88.4 | 3.2 | H | 95.9 | 3.2 | −1.64 | Reduced stability |
| Q151K | H | 58.4 | 3.4 | H | 72.1 | 3.3 | −0.44 | Reduced stability |
| D154E | H | 45.9 | 3.9 | H | 51.1 | 3.8 | 1.25 | Increased stability |
| V170I | E | 1.5 | 6.8 | E | 1.4 | 7.5 | −0.57 | Reduced stability |
| L172M | H | 0.3 | 9.6 | H | 0.2 | 9.6 | −0.76 | Reduced stability |
| V175I | H | 3.3 | 7.2 | H | 1.9 | 7.3 | −0.15 | Reduced stability |
| D214N | b | 35.9 | 4.0 | b | 48.1 | 3.6 | 0.17 | Increased stability |
| L229I | a | 39.5 | 3.7 | a | 35.1 | 3.8 | −0.5 | Reduced stability |
| V253I | H | 10.8 | 7.6 | H | 9.1 | 7.8 | −0.2 | Reduced stability |
| R255Q | H | 69.1 | 3.4 | H | 85.8 | 3.4 | 0.15 | Increased stability |
| D263E | a | 76.3 | 3.5 | a | 89.0 | 3.3 | 0.06 | Increased stability |
| Q265H | g | 91.6 | 3.3 | g | 95.3 | 3.2 | 0.18 | Increased stability |
| S286T | g | 99.6 | 3.2 | b | 87.9 | 3.6 | 0.0 | Increased stability |
| V296M | H | 6.6 | 5.4 | H | 6.4 | 5.5 | −0.7 | Reduced stability |
| L331Q | H | 0.0 | 8.7 | H | 0.2 | 8.2 | −1.92 | Reduced stability |
| E333Q | H | 66.9 | 3.3 | H | 71.3 | 3.3 | −0.39 | Reduced stability |
| M353L | b | 0.3 | 6.8 | p | 0.3 | 6.5 | 0.23 | Increased stability |
| L366I | H | 3.9 | 5.8 | H | 4.0 | 6.0 | −0.51 | Reduced stability |
| K422N | t | 83.2 | 3.2 | t | 106.0 | 3.3 | 0.54 | Increased stability |
| K424N | H | 58.1 | 3.5 | H | 65.2 | 3.6 | −0.86 | Reduced stability |
| S437T | b | 21.6 | 3.8 | b | 23.0 | 4.1 | 0.23 | Increased stability |
| M450N | H | 2.8 | 6.9 | H | 5.0 | 7.3 | −2.42 | Reduced stability |
| L456I | H | 0.2 | 13.1 | H | 1.7 | 12.8 | −0.51 | Reduced stability |
| I457V | H | 0.0 | 10.1 | H | 0.0 | 10.2 | −2.48 | Reduced stability |
| l479F | a | 67.2 | 3.4 | a | 74.9 | 3.4 | −0.09 | Reduced stability |
For the further assessment of mutations, we have used the CUPSAT server which gives a result that a mutation is stabilizing or destabilizing and favorable or unfavorable. CUPSAT server predicted all the selected stabilized mutations from the SDM server shows that the effect of mutation is unfavorable even though having stability and few mutations are destabilizing and unfavorable but only three residues H65R, D154E, and K422N have shown stabilizing and favorable effect. Table 2 shows the most destabilizing/stabilizing and favorable/unfavorable mutations along with the change in folding free (??G in kcal/mol).
Table 2: CUPSAT predicted scores.
| Mutation | SDM | CUPSAT | CUPSAT |
|---|---|---|---|
| Outcome | Predicted DDG | Outcome | |
| H54R | Increased stability | 0.03 | Stabilizing, Unfavorable |
| H65Y | Increased stability | 0.56 | Stabilizing, Favorable |
| H28R | Increased stability | 0.19 | Destabilizing, Unfavorable |
| S29T | Increased stability | −0.06 | Destabilizing Favorable |
| F46L | Increased stability | 1.34 | Stabilizing; Unfavorable |
| K55Q | Increased stability | −1.14 | Destabilizing; Unfavorable |
| F57Y | Increased stability | −1.6 | Destabilizing Favorable |
| E122Q | Increased stability | −1.43 | Destabilizing; Unfavorable; |
| D154E | Increased stability | 0.74 | Stabilizing Favorable |
| D214N | Increased stability | −0.42 | Destabilizing; Favorable; |
| R255Q | Increased stability | −0.46 | Destabilizing; Favorable |
| D263E | Increased stability | 0.26 | Stabilizing; Unfavorable; |
| Q265H | Increased stability | −0.17 | Destabilizing; Unfavorable; |
| S286T | Increased stability | −0.84 | Destabilizing; Unfavorable; |
| M353L | Increased stability | −1.52 | Destabilizing; Favorable; |
| K422N | Increased stability | 0.01 | Stabilizing Favorable |
| S437T | Increased stability | −0.54 | Destabilizing; Unfavorable |
3.2. Conservation Analysis
Using the Bayesian approach, the ConSurf server characterizes the evolutionary conservation of amino acids and their putative functional and structural profile of CYP3A4 as shown in Figure 4. A mutation in a well-conserved location may influence the function of the protein. The conservation level of amino acids at positions H65, D154, and K422 was estimated using the ConSurf tool. The ConSurf analysis predicted that mutant Histidine at position 65 was located in the variable region and found to be buried in the wild type residue, whereas D at position 154 and Lysine at position 422 are located in the vicinity of the variable region and found in the exposed region of wild type. Therefore, mutations at positions H65, D154, and K422 might have no functional impact on protein.
 | Figure 4: ConSurf color-coded sequence of CYP3A4 protein. e - An exposed residue according to the neural-network algorithm. b - A buried residue according to the neural-network algorithm. f - A predicted functional residue (highly conserved and exposed). s - A predicted structural residue (highly conserved and buried). X - Insufficient data - the calculation for this site was performed on <10% of the sequences. [Click here to view] |
3.3. Mutants Model Construction and Validation
The mutant protein structure prediction is vital to interpret how the substitution of an amino acid can alter the protein structural characteristics. The CYP3A4 protein mutant models with substitutions at H65R, D154E, and K422N accomplished with the “Build Mutant” protocol using DS modeler and energy minimized by applying CHARMm force fields in DS. To determine the quality of our model, the final refined modeled structure of CYP3A4 mutants (H65R, D154E, and K422N) is investigated by the PROCHECK, PROSA, and RMSD prediction. Using Procheck, the overall stereo chemical quality of the modeled protein is evaluated by analyzing the overall residue by residue geometry and psi and phi torsion angles of Ramachandran Plot. Ramachandran’s map of the wild protein and mutant models and are shown in Figure 5 and the plot statistics are shown in Table 3.
 | Figure 5: The stereochemical quality analysis of wild type and mutant protein models by Procheck (Ramachandran plot – favored region is red in color; allowed region is yellow in color; disallowed region is white in color) and ProSa [overall model quality (black dots indicate the match between experimentally solved protein structures distinguished by dark blue (X-ray) and light blue (NMR). [Click here to view] |
Table 3: The structure validation results of wild protein and mutant models of CYP3A4.
| Structure | Favored | Additionally Allowed | Generously allowed | Disallowed | Prosa | RMSD with wild | |
|---|---|---|---|---|---|---|---|
| Wild protein (1TQN) | 84.7 | 12.6 | 2.0 | 0.7 | −9.46 | All atoms | c-alpha |
| H65R mutant model | 83.0 | 13.8 | 2.0 | 1.2 | −9.04 | 0.15 | 0.11 |
| D154E mutant model | 84.5 | 12.8 | 2.0 | 0.7 | −9.54 | 0.16 | 0.18 |
| K422N mutant model | 84.5 | 12.6 | 2.0 | 1.0 | −9.47 | 0.09 | 0.08 |
Further validation is done by ProSA which gives the Z-score value for the comparison of compatibility. Quality assessment of the model via ProSA revealed that the mutant modeled structures matched with the NMR region of the plot with Z scores which are reliable to the Z-score of the wild type (−9.00). It signifies the quality of our model. The ProSA Z-scores of the mutant models of CYP3A4 are −9.04 (Mutant H65R), −9.54 (Mutant D154E), and −9.47 (Mutant K422N) [Figure 5 and Table 3].
To estimate the RMSD in mutated models of CYP3A4, superimposition of the C-alpha traces and main chain atoms with the wild protein (1TQN) was done using SPDBV as shown in Table 3. RMSD value is a measurable metric of structural closeness between two atomic coordinates when overlapped on each other. The C-alpha traces or main chain demonstrate the RMSD values at intervals of 0 and <2.0 Å. The greater the RMSD value of two query structures implies their dissimilarity and zero means they are similar in structure. The RMSD values at C-alpha traces and main chain atoms calculated are found to be 0.15 Ao (Mutant H65R), 0.16 Ao (Mutant D154E), and 0.09 (Mutant K422N), respectively, indicates close homology with the wild type CYP3A4. This analysis highlights the profound structural influences determined by replaced amino acid residues in the wild CYP3A4 protein molecule structure.
3.4. Docking Analysis
Molecular docking studies were accomplished using DS LibDock to explore the binding affinity of wild and mutants with the taken ligands. The compounds and the protein structures were at first energy minimized with the addition of the CHARMm force field in DS. With this energy minimized compounds, molecular docking with both wild and mutant forms of CYP3A4 was carried out to assess their comparative inhibitory properties. To evaluate the ligand-binding affinity, the LibDock score is considered. In addition, the other input docking parameters are further investigated for assessing the docking efficaciousness of the ligand with the wild and mutant proteins in our study. The best-docked conformations with a high dock score and lowest binding energy were selected for further analysis. From the results as shown in Table 4, it was revealed that quinidine showed the high dock score among all the docked compounds against both wild and mutant proteins. The stability assessment of the docked poses of quinidine with the best score was estimated by concluding the HB between the wild and mutant proteins with the ligand.
Table 4: Docking results of wild and mutated CYP3A4.
| Name of the compound | Wild 1TQN | H65Y | D154E | K422N | ||||
|---|---|---|---|---|---|---|---|---|
| LibDock Score | Binding energy | LibDock Score | Binding energy | LibDock Score | Binding energy | LibDock Score | Binding energy | |
| Quinidine | 87.126 | −31.19657 | 121.907 | 85.35163 | 121.658 | 101.32864 | 134.605 | −50.14959 |
| Nifedipine | 76.157 | −35.92159 | 115.883 | 191.76695 | 95.519 | 356.302 | 92.584 | 417.32038 |
| Midazolam | 79.095 | −25.38611 | 101.281 | 28.53686 | 93.729 | 7.66022 | 116.119 | 214.11604 |
| imipramine | 80.777 | −13.74076 | 96.692 | 224.98774 | 95.519 | 356.302 | 121.565 | 149.37450 |
The quinidine was determined to release the binding energy (DG) of −31.19 kcal/Mol with a docking score of 87.126 forming only one HB with the ARG375 residue of CYP3A4 wild type [Table 5]. The docking score of quinidine with mutated CYP3A4 (H65RR) is 121.907 with a binding energy of 85 kcal/Mol and forms 2 HBs with ARG375 and ILE443 residues. D154E mutant form of CYP3A4 interacts with quinidine by forming only one HB with ILE443 amino acid with a docking score and binding energy of 121.658 and 101.32864 kcal/Mol. Whereas, with K422L mutant form of CYP3A4, quinidine forms two HBs with ARG440 and ARG105 amino acid with a docking score and binding energy of 134.605 and -50.14959 kcal/Mol. Hence, all the mutated forms of CYP3A4 have shown a better binding affinity with quinidine among all other compounds with high docking scores than wild CYP3A4 protein. These results show that the mutated forms of CYP3A4 show better activity than the wild type. Of all the mutated forms, K422N mutated CYP3A4 shown a higher binding affinity with quinidine with high dock score compared to (87.126), H65Y (121.907), and D154E (121.658). However, from the analysis, it was found that a single HB interaction was found with wild type CYP3A4, whereas two HB interactions are formed with mutants H65Yand K422N and a single HB interaction was found with D154E. The HB interactions ensure that a ligand molecule binding to the protein structure remains in a stable conformation, which impacts the ligand’s activity. Hence, the mutant K422N having stable conformation than wild and other mutant’s forms of CYP3A4. However, it was found that the mutant K422N was having a HB distances of 2.497000 Å and 2.483000 Å with two amino acids ARG440 and ARG105, respectively. In each of the amino acid interactions with the ligand, the same oxygen atom (O2) acts as an electron acceptor and the hydrogen atoms of each of ARG440 and ARG105 acts as an electron donor. Furthermore, the HB distance between the two atoms was should be shorter, the shorter the distance, the stronger the formed HB interactions. The HB distances of K422N mutant (ARG440 - 2.497000 Å and ARG105 -2.483000 Å) were reliable, as no longer distances were found.
Table 5: The visualization of molecular docking analysis of wild type and mutant forms of CYP3A4 molecule against quinidine and their interactions and H-bond distances.
| Structure | Quinidine | Interacting atoms | H-bonddistance |
|---|---|---|---|
| Wild 1TQN | 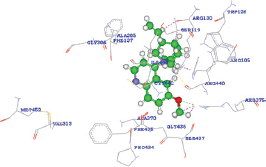 | A: ARG375:HH12 - Quinidine: O2 | 2.024000 |
| A: ARG375:HH12 - Quinidine: C24 | 2.006000 | ||
| A: ARG375:HH12 - Quinidine: H48 | 1.431000 | ||
| A: ARG105:HH22 - Quinidine: H27 | 1.809000 | ||
| A: ARG440:O - Quinidine: H35 | 2.014000 | ||
| H65R | 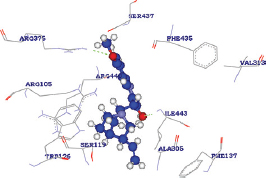 | A: ARG375:HH22 - Quinidine: O2 | 2.447000 |
| A: ILE443:HN - Quinidine: O1 | 2.363000 | ||
| D154E | 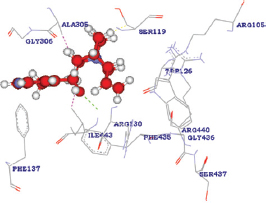 | A: ILE443:HN - Quinidine: O1 | 2.283000 |
| Quinidine: H25 - A: ILE443:CG2 | 2.147000 | ||
| Quinidine: H27 - A: ALA305:CB | 1.680000 | ||
| Quinidine: H31 - A: SER119:CB | 2.015000 | ||
| K422N | 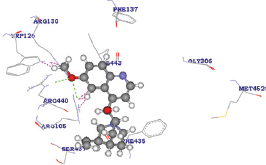 | A: ARG440:HE - Quinidine: O2 | 2.497000 |
| A: ARG105:HH22 - Quinidine: O2 | 2.483000 | ||
| Quinidine: H48 - A: TRP126:NE1 | 1.629000 | ||
| Quinidine: H42 - A: ARG105:NH2 | 1.687000 |
The mutant K422N and quinidine complex showed lower binding energy (−50.14959 kcal/mol) than the wild type and other mutants of CYP3A4. Thus, the mutation K422N corroborated to a to a stronger enzyme-substrate complex. Whereas, the docked complexes of quinidine with the CYP3A4 wild (−31.19657) indicates a stronger bond and with H65Y (85.35163) and D154E (101.32864) corresponds to a weaker bonding.
The overall alterations in HB at the amino acid substitution site could be the reason behind the changes in the binding affinity. Hence, considering the overall analysis mutant K422N was found to the most favorable in all terms of docking score, HB interactions and their distances with least binding energy.
4. CONCLUSION
In this study, analysis of stability and binding efficacy of CYP3A4 mutants were carried out through various in silico methods. The analysis predicted the substituted K422N to be more favorable in terms of stability and have shown higher binding affinity with the taken compound compared to wild CYP3A4. Our results demonstrate that these mutational studies give an insight for guidelines for performing the functional assays of the mutant proteins and might be used as an important tool for benefiting further in vitro studies.
5. ACKNOWLEDGEMENT
The authors express gratitude to SERB-YSS (YSS/2015/000173) program of Ministry of Science and Technology and RUSA 2.0 program of Ministry of Human Resource Development, Govt. of India for their financial support.
6. AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
7. FUNDING
SERB-YSS (YSS/2015/000173) program, Ministry of Science and Technology, Govt. of India. RUSA 2.0 program, Ministry of Human Resource Development, Govt. of India.
8. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
9. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
10. DATA AVAILABILITY
The raw data supporting this article will be made available by the authors, without reservation.
11. PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Esteves F, Rueff J, Kranendonk M. The central role of cytochrome P450 in xenobiotic metabolism-a brief review on a fascinating enzyme family. J Xenobiot 2021;11:94-114. [CrossRef]
2. Guengerich FP. Intersection of the roles of cytochrome P450 enzymes with xenobiotic and endogenous substrates:Relevance to toxicity and drug interactions. Chem Res Toxicol 2017;30:2-12. [CrossRef]
3. Spicakova A, Bazgier V, SkálováL, Otyepka M, Anzenbacher P. b-caryophyllene oxide and trans-nerolidol affect enzyme activity of CYP3A4-in vitro and in silico studies. Physiol Res 2019;68:51-8. [CrossRef]
4. Guengerich FP. A history of the roles of cytochrome P450 enzymes in the toxicity of drugs. Toxicol Res 2021;37:1-23. [CrossRef]
5. Niwa T, Okamoto A, Narita K, Toyota M, Kato K, Kobayashi K, et al. Comparison of steroid hormone hydroxylation mediated by cytochrome P450 3A subfamilies. Arch Biochem Biophys 2020;682:108283. [CrossRef]
6. Jackson KD, Durandis R, Vergne MJ. Role of cytochrome P450 enzymes in the metabolic activation of tyrosine kinase inhibitors. Int J Mol Sci 2018;19:2367. [CrossRef]
7. Sevrioukova IF, Poulos TL. Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc Natl Acad Sci U S A 2017;114:486-91. [CrossRef]
8. Deodhar M, Al Rihani SB, Arwood MJ, Darakjian L, Dow P, Turgeon J, et al. Mechanisms of CYP450 inhibition:Understanding drug-drug interactions due to mechanism-based inhibition in clinical practice. Pharmaceutics 2020;12:846. [CrossRef]
9. Kumondai M, Rico EM, Hishinum E, Ueda A, Saito S, Saigusa D, et al. Functional characterization of 40 CYP3A4 variants by assessing midazolam 1-hydroxylation and testosterone 6b-hydroxylation. Drug Metab Dispos 2021;49:212-20. [CrossRef]
10. Zhou XY, Hu XX, Wang CC, Lu XR, Chen Z, Liu Q, et al. Enzymatic activities of CYP3A4 allelic variants on quinine 3-hydroxylation in vitro. Front Pharmacol 2019;10:591. [CrossRef]
11. Arendse LB, Blackburn JM. Effects of polymorphic variation on the thermostability of heterogenous populations of CYP3A4 and CYP2C9 enzymes in solution. Sci Rep 2018;8:1-14. [CrossRef]
12. Gaedigk A, Sundberg MI, Miller NA, Leeder JS, Carrillo MW, Klein TE. The pharmacogene variation (PharmVar) consortium:Incorporation of the human cytochrome p450 (CYP) allele nomenclature database. Clin Pharmacol Ther 2018;103:399-40. [CrossRef]
13. Guttman Y, Nudel A, Kerem Z. Polymorphism in cytochrome P450 3A4 is ethnicity related. Front Genet 2019;10:224. [CrossRef]
14. Fang P, Tang PF, Xu RA, Zheng X, Wen J, Bao SS, et al. Functional assessment of CYP3A4 allelic variants on lidocaine metabolism in vitro. Drug Des Dev Ther 2017;11:3503-10. [CrossRef]
15. Hu GX, Dai DP, Wang H, Huang XX, Zhou XY, Cai J, et al. Systematic screening for CYP3A4 genetic polymorphisms in a Han Chinese population. Pharmacogenomics 2017;18:369-79. [CrossRef]
16. Yong CC, Zheng X, Liu TH, Wang CC, Tang PF, Chen Z, et al. Functional characterization of 21 CYP3A4 variants on amiodarone metabolism in vitro. Xenobiotica 2019;49:120-6. [CrossRef]
17. Quiroga I, Scior T. Induced fit for cytochrome P450 3A4 based on molecular dynamics. ADMET DMPK 2019;7:252-66. [CrossRef]
18. Bawono P, Dijkstra M, Pirovano W, Feenstra A, Abeln S, Heringa J. Multiple sequence alignment. In:Methods in Molecular Biology. New York:Humana Press;2017. 167-89. [CrossRef]
19. Pandurangan AP, Montaño BO, Ascher DB, Blundell TL. SDM:A server for predicting effects of mutations on protein stability. Nucleic Acids Res2017;45:W229-35. [CrossRef]
20. Sanavia T, Birolo G, Montanucci L, Turina P, Capriotti E, Fariselli P. Limitations and challenges in protein stability prediction upon genome variations:Towards future applications in precision medicine. Comput Struct Biotechnol J 2020;18:1968-79. [CrossRef]
21. Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. Consurf 2016:An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 2016;44:W344-50. [CrossRef]