
1. INTRODUCTION
Bats act as a natural host for a number of emerging viruses that can cause disease in humans, including RNA viruses such as Nipah, Marburg, Sosuga, and Hendra [1-4]. Ebola, Middle East Respiratory Coronavirus, Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), and SARS-CoV2 are important viruses that might have originated in bats, despite the fact that other hosts do exist. A recent study has revealed that the number of viral variants is proportional to the number of virus species [5]. As compared to other mammalian orders, bats have a higher viral variety and indeed the largest number of mammalian species is found in Chiroptera (bats) and Rodentia (rodents). The viral variety of bats makes them an essential taxonomic group for global virus detection and zoonotic disease monitoring [6].
The Rhabdoviridae RNA virus family is one of the most ecologically diverse viral families [7,8]. Rhabdoviruses might well be found in a wide range of birds, reptiles, mammals, and fish, as well as in plants and other animals, and some of them are spread by arthropod vectors. A member of this family, the rabies virus, is responsible for approximately 25,000 human deaths each year [9]. The non-segmented ssRNA genome of rhabdoviruses is packed inside a bullet or rod-like particle that carries five structural proteins, namely matrix protein, glycoprotein, nucleoprotein, polymerase-related phosphoprotein, and RNA-based RNA polymerase [10]. The genome contains five ORFs ordered in the pattern 3’-N-P-M-G-L-5’, along with partially complementary, untranslated leader (l) and trailer (t) sequences. The five matching capped and polyadenylated mRNAs are expressed by means of the relatively conserved transcription initiation (TI) and transcription termination/polyadenylation (TTP) sequences that border each ORF [10]. Other ORFs in rhabdovirus genomes have been discovered to encode hypothetical proteins, the majority of which have no known function and can exist as alternative ORFs overlapping within protein gene structure [8]. On the other hand, ORFs may be flanked by TTP or TI sequences positioned between the structures of protein genes, some of which appear to have evolved as a result of gene duplication [8,11-14].
Codon usage bias (CUB) is a widely used method for determining the elements that govern viral evolution [15]. The CUB of the viral gene might be associated with a particular host selection, leading to a deeper understanding of the host’s adaptive response to the evolution of a virus and infection [16]. CUB refers to the widespread phenomenon of synonymous codons being used at different frequencies [17,18]. Synonymous codons, on the other hand, are not employed randomly and some are consistently chosen over others when encoding an amino acid. The frequency of synonymous codons differed not just between genomes but also between functionally related genes and within the limits of a single gene [19,20]. CUB was also affected by GC-biased gene conversion and GC heterogeneity, which was determined by the degree of local recombination [21-23]. Previous studies have revealed that synonymous codon usage might have a major impact on genome evolution due to the influence of natural selection, mutation pressure, and genetic drift on gene translational efficiency [21-23].
In addition to protein structure, function, and translational folding in tandem, CUB has an impact on a wide range of biological functions, including mRNA stability, translation efficiency, and transcription [18,24,25]. CUB was found to affect translation efficiency and transcription rates by altering mRNA folding and chromatin formation, as well as translation elongation rate, suggesting that it was the consequence of genomic adaptation to transcription and translation processes [24,26]. CUB analysis revealed that evolutionary linkages and horizontal gene transfers across animals may be discovered because codon usage patterns are comparable in the closely related taxa [25]. The main objective of this study was to analyze the nucleotide frequency and CUB pattern in 15 Rhabdoviridae families, as well as the impact of synonymous codon usage in the evolutionary processes of rhabdovirus. We analyzed the codon usage pattern in the Rhabdoviridae family to understand the driving mechanisms of CUB using different codon usage indices, such as genomic GC contents in the 1st, 2nd, and 3rd codon positions, neutrality plot, parity rule-2 (PR2) plot, translational selection, nucleotide skewness, and relative synonymous codon usage (RSCU). More particular information regarding virus evolution can be gleaned through codon usage patterns and pathogenicity, which can assist in vaccine development with greater efficacy and strengthen the control efforts to prevent the virus-borne zoonosis from spreading.
2. MATERIALS AND METHODS
2.1. Retrieval of Sequence Data
We downloaded the coding sequences (CDS) of 15 different viral genomes of the Rhabdoviridae family, namely West Caucasian bat virus, European bat Lyssavirus 1, Shimoni bat virus, Lagos bat virus, European bat lyssavirus 2, Kolente virus isolate K7292, Australian bat lyssavirus, Fikirini bat rhabdovirus, Irkut virus, Aravanvirus, Mount Elgon bat virus, Bokeloh bat lyssavirus 21961, American bat vesiculo virus, Khujand lyssavirus, and Kumasi rhabdovirus from the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov). In this study, we considered only the CDS that was exact multiples of three nucleotides and had valid start and stop codons, excluding any unidentified bases.
2.2. Nucleobase Composition
In this study, we analyzed the overall nucleotide compositions (A%, G%, T%, and C%) and its compositions at the third codon positions (A3%, G3%, T3%, and C3%) of each viral CDS. The total GC contents at the 1st (GC1%), 2nd (GC2%), and 3rd (GC3%) synonymous codon positions were also determined. Then, we computed the nucleotide skewness, such as GC skew, AT skew, purine skew, pyrimidine skew, keto skew, and amino skew values of CDS over all genomes.
2.3. RSCU
RSCU is the proportion of the observed incidence of a codon to its random incidence when all the synonymous codons of an amino acid are used equally. A codon’s RSCU value >1 indicates that it occurs more frequently in the CDS, whereas a codon’s RSCU value <1 denotes that it happens much less often [27]. RSCU value >1.6 denotes that a codon is over-represented in a CDS, whereas RSCU <0.6 denotes that a codon is under-represented [28].
2.4. Effective Number of Codons (ENC)
ENC is a good parameter for estimating the CUB in CDS, regardless of the gene length or the number of amino acids in the encoded protein. It refers to how diversified the codon usage of a gene is, as opposed to how uniformly synonymous codons are utilized. The ENC value ranges from 20 (when only one amino acid is encoded by a single codon) up to 61 (when all synonymous codons for different amino acids are randomly used). Significant CUB is indicated by an ENC value <35 [29].
ENC formula:
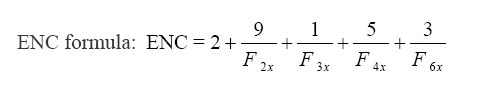
Where, Fk stands for the mean value for k-fold degenerate amino acids with Fkx (k = 2, 3, 4, or 6 depending on degeneracy level). When two codons of the same amino acid are randomly picked and found to be identical, the F value is calculated [29].
2.5. PR2 Plot
In PR2 plot analysis, GC bias, i.e., (G3/[G3 + C3]) and AT bias, i.e., (A3/[A3 + T3]) were plotted in the abscissa and ordinate, respectively. It is employed to assess the impact of the forces of evolution (i.e., selection and mutational pressure) [30]. Here, 0.5 denotes the center point of the graph, indicating that there is neither selection nor mutational bias in the two strands of the complementary DNA.
2.6. Correspondence Analysis (COA)
COA is a multivariate tool based on RSCU values of codons to evaluate the trends of codon usage patterns in the CDSs [31]. The COA graph was created using RSCU data consisting of 59 synonymous codons encoding 18 amino acids (except Met and Trp). To investigate the patterns in codon usage variation, the researchers can use the idea of relative inertia, in which genes occupy a specified position in a graph. The gene’s position was investigated to learn more about the factors that have an impact on the codon usage patterns.
2.7. Neutrality Plot
To identify the prevailing impact of evolutionary forces on CUB, a neutrality plot is commonly used. The graph was generated by plotting the X-axis with GC3 and the Y-axis with GC12. In the graphical representation, a slope (regression coefficient [RC]) close to 0 implies that directional mutation pressure has no effect (natural selection plays a dominant role), whereas a slope close to 1 indicates total neutrality (mutation pressure plays a dominant role) [32].
2.8. Translational Selection (P2) and Mutational Responsive Index (MRI)
P2 is a measure of the codon’s ability to interact with the specific anticodon and is related to gene translation precision. According to Gouy and Gautier (1982) [33], if P2 value is larger than 0.5 denotes that the CDS is affected by translational selection [33].
The degree of mutational drift is indicated by the MRI value of a CDS [34]. A positive value of MRI suggests that mutations have a considerable impact on the CDSs, whereas a negative value shows that translational selection has a large impact CDS [34].
2.9. Phylogenetic Study
The complete CDSs of 15 different viral genomes of the Rhabdoviridae family were obtained from NCBI database in FASTA format. To comprehend the evolutionary link, a phylogenetic tree was created using these sequences. The phylogenetic tree was created using MEGA11. The maximum likelihood technique based on the Tamura-Nei model was used to infer the past evolutionary history [35,36].
2.10. Software
CUB indices of the Rhabdoviridae family were calculated using a PERL-based computer program developed by the corresponding author (SC). The COA was performed using the Paleontological Statistics Software program (PAST) [37] to evaluate the changes in codon usage. To estimate the dN/dS value for Rhabdoviridae family members, we used the SLAC method in Datamonkey [38]. The phylogenetic tree was created using MEGA 11 software based on the maximum likelihood approach [39]. A consensus tree based on bootstrapping derived from 1000 iterations was used to depict the history of the evolution of the Rhabdoviridae family. All statistical analyses including correlation analysis were performed using IBM SPSS software, version 21.0 [40].
3. RESULTS AND DISCUSSION
3.1. Compositional Properties
The base composition has a large influence on the CUB, and it could be the most essential component in organizing the codon usage patterns [15]. We studied the nucleobase composition of CDS in the Rhabdoviridae family [Figures 1 and 2]. Among the four nucleotides, A% was determined to be the most abundant. Our result is comparable to the study on nucleotide composition in mouse mammary tumor virus and astrovirus, which found similar patterns, with A% being the highest composition [41]. In Rhabdoviridae, a higher AT% relative to GC% indicated AT-richness. AT-rich was found in hepadnavirus genes, which is compatible with our results [42]. AT content causes low thermodynamic stability during replication [43]. Hence, we hypothesized that the higher AT content of the Rhabdoviridae might have played a role in the mRNA stability during replication.
 | Figure 1: Overall nucleotide composition and nucleotide composition at the 3rd codon position found to be AT-rich (as GC value is 44.96 and GC3% is 45.48) in Rhabdoviridae. [Click here to view] |
 | Figure 2: GC content (GC1%>GC2%>GC3%) at all the three codon positions of Rhabdoviridae. [Click here to view] |
GC% at all three codon positions, i.e., GC1, GC2, and GC3 were calculated and found GC1>GC3>GC2. A substantial component influencing CUB across genomes has been revealed to be GC composition [44]. At the third codon position, T3% was found to be the highest, followed by A3%, C3% and G3%. Similarly, in hepadnavirus, the nucleotide compositions at the third codon position were followed by T3%>A3%>C3%, and G3% [42]. The correlation of compositional features can be used to discover the major elements influencing CUB [45]. The overall composition of nucleobases, A%, T%, G%, and C% correlated to the nucleobase composition at the codons wobble location, A3%, T3%, G3%, and C3 % using Karl Pearson’s approach [Table 1]. We found a significant correlation between the corresponding nucleotide bases, indicating that mutational pressure was exerted on the CDS of Rhabdoviridae for creating CUB.
Table 1: Interrelationships of overall base composition with base composition at the third codon position.
| A3% | T3% | G3% | C3% | GC3% | |
|---|---|---|---|---|---|
| A% | 0.771** | 0.175 | −0.285* | −0.695** | −0.599** |
| T% | 0.271* | 0.751** | −0.613** | −0.448** | −0.649** |
| G% | −0.586** | −0.501** | 0.790** | 0.334** | 0.688** |
| C% | −0.529** | −0.538** | 0.259* | 0.849** | 0.677** |
| GC% | −0.717** | −0.670** | 0.643** | 0.795** | 0.880** |
3.2. Immensity of CUB
To establish the level of CUB in Rhabdoviridae, the ENC values were calculated and found that the values ranged from 40.8 to 60.0 [Table 2]. Rhabdoviridae had a mean ENC value of 53.64 (i.e., larger than 35), indicating a low CUB of the genes. Another study revealed low CUB in 50 human RNA viruses, with ENC values ranging from 38.9 (hepatitis A virus) to 58.3 (eastern equine encephalitis virus) [15], which is consistent with our findings. The ENC values of SARS-CoV ranged from 42.19 to 59.06, with a mean of 48.99, indicating that the CUB in the SARS-CoV genome is low [46].
Table 2: ENC, P2, and MRI values of different genes in Rhabdoviridae.
| Accession No. | Virus | ENC value | Translational selection (P2) value | MRI value |
|---|---|---|---|---|
| NC_025377.1 | West Caucasian bat virus, nucleoprotein gene | 52.00 | 0.05 | 0.42 |
| NC_025377.1 | West Caucasian bat virus, phosphoprotein gene | 48.90 | 0.08 | 0.59 |
| NC_025377.1 | West Caucasian bat virus, matrix protein gene | 54.30 | 0.14 | 0.63 |
| NC_025377 | West Caucasian bat virus, glycoprotein gene | 51.80 | 0.04 | 0.56 |
| NC_025377.1 | West Caucasian bat virus, polymerase gene | 52.30 | 0.01 | 0.53 |
| NC_025365.1 | Shimoni bat virus, nucleoprotein gene | 47.10 | 0.05 | 0.51 |
| NC_025365.1 | Shimoni bat virus, phosphoprotein gene | 51.30 | 0.08 | 0.55 |
| NC_025365.1 | Shimoni bat virus, matrix protein gene | 51.50 | 0.13 | 0.58 |
| NC_025365.1 | Shimoni bat virus, glycoprotein gene | 49.50 | 0.04 | 0.48 |
| NC_025365.1 | Shimoni bat virus, polymerase protein gene | 46.90 | 0.01 | 0.48 |
| NC_020807.1 | Lagos bat virus isolate 0406SEN, nucleoprotein gene | 50.20 | 0.05 | 0.47 |
| NC_020807.1 | Lagos bat virus isolate 0406SEN, phosphoprotein gene | 48.60 | 0.09 | 0.45 |
| NC_020807.1 | Lagos bat virus isolate 0406SEN, matrix protein gene | 55.10 | 0.14 | 0.56 |
| NC_020807.1 | Lagos bat virus isolate 0406SEN, glycoprotein gene | 55.90 | 0.04 | 0.51 |
| NC_020807.1 | Lagos bat virus isolate 0406SEN, polymerase gene | 53.00 | 0.01 | 0.49 |
| NC_009528.2 | European bat lyssavirus 2 isolate RV1333, nucleoprotein gene | 50.60 | 0.05 | 0.54 |
| NC_009528.2 | European bat lyssavirus 2 isolate RV1333, phosphoprotein gene | 58.90 | 0.09 | 0.66 |
| NC_009528.2 | European bat lyssavirus 2 isolate RV1333, matrix protein gene | 54.10 | 0.14 | 0.58 |
| NC_009528.2 | European bat lyssavirus 2 isolate RV1333, glycoprotein gene | 56.10 | 0.04 | 0.51 |
| NC_009528.2 | European bat lyssavirus 2 isolate RV1333, L protein gene | 53.90 | 0.01 | 0.55 |
| NC_009527.1 | European bat lyssavirus 1, nucleoprotein gene | 52.40 | 0.05 | 0.48 |
| NC_009527.1 | European bat lyssavirus 1, phosphoprotein gene | 53.90 | 0.10 | 0.53 |
| NC_009527.1 | European bat lyssavirus 1, matrix protein gene | 50.10 | 0.15 | 0.54 |
| NC_009527.1 | European bat lyssavirus 1, glycoprotein gene | 51.10 | 0.05 | 0.64 |
| NC_009527.1 | European bat lyssavirus 1, L protein gene | 53.10 | 0.01 | 0.50 |
| NC_025342.1 | Kolente virus isolate DakAr K7292, N protein gene | 56.10 | 0.06 | 0.58 |
| NC_025342.1 | Kolente virus isolate DakAr K7292, P protein gene | 60.00 | 0.09 | 0.66 |
| NC_025342.1 | Kolente virus isolate DakAr K7292, M protein gene | 60.00 | 0.12 | 0.53 |
| NC_025342.1 | Kolente virus isolate DakAr K7292, G protein gene | 57.40 | 0.04 | 0.43 |
| NC_025342.1 | Kolente virus isolate DakAr K7292, L protein gene | 56.00 | 0.01 | 0.51 |
| NC_025341.1 | Fikirini bat rhabdovirus isolate KEN352, nucleocapsid gene | 58.60 | 0.06 | 0.53 |
| NC_025341.1 | Fikirini bat rhabdovirus isolate KEN352, phosphoprotein gene | 56.50 | 0.09 | 0.50 |
| NC_025341.1 | Fikirini bat rhabdovirus isolate KEN352, matrix protein gene | 56.00 | 0.13 | 0.68 |
| NC_025341.1 | Fikirini bat rhabdovirus isolate KEN352, glycoprotein gene | 57.00 | 0.04 | 0.51 |
| NC_025341.1 | Fikirini bat rhabdovirus isolate KEN352, RNA-dependent RNA polymerase gene | 55.80 | 0.01 | 0.55 |
| NC_020809.1 | Irkut virus, nucleoprotein gene | 50.80 | 0.05 | 0.49 |
| NC_020809.1 | Irkut virus, phosphoprotein gene | 54.70 | 0.09 | 0.44 |
| NC_020809.1 | Irkut virus, matrix protein gene | 52.50 | 0.14 | 0.60 |
| NC_020809.1 | Irkut virus, glycoprotein gene | 53.00 | 0.05 | 0.45 |
| NC_020809.1 | Irkut virus, polymerase gene | 53.40 | 0.01 | 0.52 |
| NC_020808.1 | Aravan virus, nucleoprotein gene | 54.30 | 0.05 | 0.52 |
| NC_020808.1 | Aravan virus, phosphoprotein gene | 55.80 | 0.09 | 0.62 |
| NC_020808.1 | Aravan virus, matrix protein gene | 54.00 | 0.14 | 0.56 |
| NC_020808.1 | Aravan virus, glycoprotein gene | 54.00 | 0.05 | 0.56 |
| NC_020808.1 | Aravan virus, polymerase gene | 53.30 | 0.01 | 0.54 |
| NC_003243.1 | Australian bat lyssavirus, nucleocapsid protein gene | 57.10 | 0.05 | 0.54 |
| NC_003243.1 | Australian bat lyssavirus, phosphoprotein gene | 57.20 | 0.09 | 0.56 |
| NC_003243.1 | Australian bat lyssavirus, matrix protein gene | 56.20 | 0.12 | 0.50 |
| NC_003243.1 | Australian bat lyssavirus, glycoprotein gene | 53.60 | 0.04 | 0.60 |
| NC_003243.1 | Australian bat lyssavirus, L protein gene | 52.70 | 0.01 | 0.50 |
| NC_034545.1 | Mount Elgon bat virus nucleoprotein, phosphoprotein, matrix, glycoprotein, and polymerase genes | 46.20 | 0.05 | 0.53 |
| NC_034545.1 | Mount Elgon bat virus nucleoprotein, phosphoprotein, matrix, glycoprotein, and polymerase genes | 50.30 | 0.08 | 0.39 |
| NC_034545.1 | Mount Elgon bat virus nucleoprotein, phosphoprotein, matrix, glycoprotein, and polymerase genes | 40.80 | 0.12 | 0.43 |
| NC_034545.1 | Mount Elgon bat virus nucleoprotein, phosphoprotein, matrix, glycoprotein, and polymerase genes | 48.30 | 0.04 | 0.46 |
| NC_034545.1 | Mount Elgon bat virus nucleoprotein, phosphoprotein, matrix, glycoprotein, and polymerase genes | 46.50 | 0.01 | 0.41 |
| NC_025251.1 | Bokeloh bat lyssavirus isolates 21961, nucleoprotein gene | 53.10 | 0.05 | 0.50 |
| NC_025251.1 | Bokeloh bat lyssavirus isolates 21961, phosphoprotein gene | 51.00 | 0.09 | 0.60 |
| NC_025251.1 | Bokeloh bat lyssavirus isolates 21961, matrix protein gene | 53.50 | 0.15 | 0.57 |
| NC_025251.1 | Bokeloh bat lyssavirus isolates 21961, glycoprotein gene | 55.70 | 0.05 | 0.53 |
| NC_025251.1 | Bokeloh bat lyssavirus isolates 21961, polymerase gene | 53.30 | 0.01 | 0.53 |
| NC_022755.1 | American bat vesiculovirus TFFN-2013 isolate liver 2008, nucleocapsid N gene | 54.70 | 0.06 | 0.50 |
| NC_022755.1 | American bat vesiculovirus TFFN-2013 isolate liver 2008, phosphoprotein P gene | 57.50 | 0.12 | 0.51 |
| NC_022755.1 | American bat vesiculovirus TFFN-2013 isolate liver 2008, matrix protein M gene | 60.00 | 0.12 | 0.50 |
| NC_022755.1 | American bat vesiculovirus TFFN-2013 isolate liver 2008, glycoprotein G gene | 52.80 | 0.05 | 0.53 |
| NC_022755.1 | American bat vesiculovirus TFFN-2013 isolate liver 2008, polymerase L gene | 53.80 | 0.01 | 0.49 |
| NC_025385.1 | Khujand lyssavirus, nucleoprotein gene | 52.20 | 0.05 | 0.50 |
| NC_025385.1 | Khujand lyssavirus, phosphoprotein gene | 50.50 | 0.10 | 0.61 |
| NC_025385.1 | Khujand lyssavirus, matrix protein gene | 54.20 | 0.14 | 0.58 |
| NC_025385.1 | Khujand Lyssavirus, glycoprotein gene | 54.50 | 0.04 | 0.56 |
| NC_025385.1 | Khujand Lyssavirus, polymerase gene | 53.80 | 0.01 | 0.48 |
| NC_028236.1 | Kumasi rhabdovirus, N gene | 53.90 | 0.06 | 0.49 |
| NC_028236.1 | Kumasi rhabdovirus, hypothetical protein 2 gene | 60.00 | 0.77 | 0.38 |
| NC_028236.1 | Kumasi rhabdovirus, hypothetical protein 3 gene | 60.00 | 2.44 | 0.33 |
| NC_028236.1 | Kumasi rhabdovirus, hypothetical protein 4 gene | 60.00 | 0.66 | 0.40 |
| NC_028236.1 | Kumasi rhabdovirus, M gene | 53.80 | 0.13 | 0.57 |
| NC_028236.1 | Kumasi rhabdovirus, G gene | 56.70 | 0.04 | 0.61 |
| NC_028236.1 | Kumasi rhabdovirus, L gene | 54.60 | 0.01 | 0.50 |
| Average | 53.64 | 0.11 | 0.52 |
ENC: Effective number of the codon, MRI: Mutational responsive index.
3.3. Role of Translational Selection (P2) and MRI
To investigate the role of CUB in mRNA translation, we calculated the P2 value. We found a mean P2 value of 0.11 across the CDS of Rhabdoviridae [Table 2]. Since the P2 value was <0.5, we could infer that translational selection might not be a prominent factor in synonymous codon usage variance [47]. Hence, translational selection might have a lesser impact on the CUB pattern of Rhabdoviridae. A study on the viral genome revealed that the CUB of structural genes should be higher than that of non-structural genes [48]. The positive value of MRI indicates that CUB is influenced by the application of directed mutational pressure, whereas the negative value of MRI indicates that translational selection has a substantial impact on CUB. To gain a better understanding of how mutational pressure affects CUB, we computed the mean values of MRI in Rhabdoviridae. The results are shown in Table 2. Here, we found a positive value of MRI, suggesting that mutational pressure might have a significant role in shaping the CUB of the Rhabdoviridae family. Our finding is comparable to the previous analysis of 11 Nipah virus genomes [49].
3.4. ENC Interrelationship with Nucleotide Skews and Base Composition
It was suggested that the composition of nucleotides significantly influences the CUB formation [15]. As a consequence, we examined the correlation between ENC and base composition at wobble positions of codons and found that A3 and ENC had a highly significant positive correlation, whereas C, GC, C3, and GC3 values had highly significant negative correlation with ENC [Table 3]. Further, we used Karl Pearson’s method for correlation analysis between ENC (as a magnitude of CUB) and nucleotide skews [Table 4]. Here, we found a highly significant negative correlation between CUB and GC skew (r = −0.344, P < 0.01), pu-skew (r = −0.462, P < 0.01), py-skew (r = −0. 681, P < 0.01), ko-skew (r = −0. 475, P < 0.01) and amino skew (r = −0.593, P < 0.01) showing that all of these skews may alter the CUB of genes across genomes. However, we found a positive correlation between ENC and AT-skew (r = 0.173) [Table 4]. The substantial relationship between ENC and nucleotide content in the first and third base positions showed that the CUB could be structured by mutational force. The correlation studies between ENC and nucleotide composition revealed a significant correlation in Nipah virus (NiV B3 and NiV USA), implying that base skewness might have a substantial impact on Nipah virus codon usage patterns [49].
Table 3: Interrelationships of ENC value with overall base composition and base composition at the third codon position.
| A% | T% | G% | C% | GC% | A3% | T3% | G3% | C3% | GC3% | |
|---|---|---|---|---|---|---|---|---|---|---|
| ENC | 1.000 | 0.009 | −0.274* | −0.727** | −0.674** | 0.771** | 0.175 | −0.285* | −0.695** | −0.599** |
Table 4: Interrelationships of ENC value with nucleotide skews.
| GC skew | AT skew | Purine skew | Pyrimidine skew | Keto skew | Amino skew | |
|---|---|---|---|---|---|---|
| ENC | −0.344** | 0.173 | −0.462** | −0.681** | −0.475** | −0.593** |
** Significant at P<0.01, ENC: Effective number of the codon.
3.5. Synonymous Codons Usage in Rhabdoviridae
The codon usage patterns of Rhabdoviridae were examined by RSCU analysis. The pattern of codons utilized in Rhabdoviridae is shown in [Figure 3]. The RSCU values mostly fell between 0.6 and 1.6, indicating a stable genetic composition. The codons TCT, AGA, and AGG have RSCU values>1.6 and were consequently overrepresented in Rhabdoviridae [Figure 4]. The underrepresented codons (i.e., RSCU value <0.6) were TCG, CCG, CGA, CGC, CGG, CGT, ACG, GTA, GCG, and GGC [Figure 4]. Further, the study of the RSCU values revealed that the T-ending codons were more common than the others. The genomes of the equine influenza virus revealed a high predilection for T-ended codons, according to RSCU analysis [50]. In avian and human influenza viruses, ACG, CCG, CGT, GCG, CGA, CGG, CGC, and TCG were the eight underrepresented codons with RSCU values <0.6, whereas ACA, AGG, AGA, GGA, and GCA were the five purine-rich overrepresented codons. From a study on the synonymous codon usage between viruses and their hosts, it was revealed that the usage of synonymous codons differed between virus and host for 14 out of 18 amino acids in the encoded proteins of both [16].
 | Figure 3: Relative synonymous codon usage (RSCU) value of codons of Rhabdoviridae (Here, RSCU values are shown on X-axis and graphs drawn on Y-axis, if the RSCU value is above 1, it indicates more frequently used, below 1 indicating less frequently used codons). [Click here to view] |
 | Figure 4: 3 Overrepresented codon, i.e., relative synonymous codon usage (RSCU) value >1.6 (AGA, AGG, and TCT) and 10 underrepresented codons, i.e., RSCU value <0.6 (TCG, CCG, CGA, CGC, CGG, CGT, ACG, GTA, GCG, and GGC). [Click here to view] |
3.6. Variation in Codon Usage
The COA revealed that the codon usage pattern varied among the viral transcriptomes. The PAST software was used to create a COA graph that depicted the trends in codon usage variance. The two primary sources of overall variation were depicted on Axis-1 and Axis-2 of the COA graph. Axis-1 accounted for 11.96% variation, whereas Axis-2 accounted for 11.27%. We illustrated a graph of the Rhabdoviridae family with bases distributed throughout the axes [Figure 5]. Several codons were found close to the axes, whereas others dispersed far away, indicating that mutation and selection pressure might alter CUB. Our findings are similar to those of the Zika virus study [51]. In a previous study, COA of viral transcriptomes revealed a significant tendency in the first axis, which accounts for 15.4% variance, as well as the fact that no other axis could contribute to more than 7.6% variation [52]. Further, in another study, the first axis of the COA graph consisting of seven viruses (H9N2, H1N1, H1N2, H1N2, H1N2, H1N2, H3N2, H5N1, H7N7, and Influenza B) revealed a prominent pattern that contributed 12.80% of the variation in general [48]. Hence, our study was comparable to these two analyses.
 | Figure 5: Correspondence analysis in Rhabdoviridae (AT-ended and GC-ended codons are represented by blue and pink dots, respectively, and distributed throughout the axes indicating that translational selection and selection pressure might alter CUB). [Click here to view] |
3.7. PR2 Plot (Parity plot)
In this study, we analyzed the PR2 bias plot to determine the impact of natural selection and mutational force in Rhabdoviridae genomes [53]. When the mutational pressure alone affects CUB, G and C (or A and T) are proportionately employed among the degenerate codon groups in a gene or genome [54]. Natural selection might not always result in the proportional usage of nucleobases (G and C or A and T). The PR2 plot is an intra-strand bias metric that shows how mutation and selection pressure affect gene codon usage [54]. In a PR2 plot, A = T and G = C (PR2) occur when both coordinates are 0.5 (center of the plot) or when there is no bias in selection pressure or mutation force in the complementary strands of DNA [30]. In this study, we used the values A3/(A3 + T3) and G3/(G3 + C3) as the ordinate and abscissa of the Rhabdoviridae’s 2-, 4- and 6-fold degenerate codons, respectively [Figure 6]. We found that the nucleotides were asymmetrically distributed across the graph, suggesting that mutation and selection pressure might have influenced the CUB of Rhabdoviridae. Furthermore, the Zika virus PR2 studies revealed that A and C bases were detected far more often, implying that both translational selection and mutation pressure drove the pattern of codon usage [51].
 | Figure 6: 6-fold, 4-fold, and 2-fold parity plots of Rhabdoviridae Bases were asymmetrically distributed across the graph indicating that translational selection and selection pressure might have influenced the CUB of Rhabdoviridae). [Click here to view] |
3.8. Natural Selection Impacts on Patterns of Codon Usage
As mutation is one the most important components in CUB, so the AT and GC content in the third codon place should be equivalent [55], however, our finding shows that at the wobble position the AT and GC composition was uneven [Figure 1]. Hence, we hypothesized that the mechanisms, such as natural selection might have a huge impact on developing the CUB of Rhabdoviridae. The presence of uneven AT and GC concentrations at the wobble position of the Nipah-virus genomes tends to support our result [49].
3.9. Neutrality Plot
The consequences of two evolutionary factors on a gene or genome are depicted in a neutrality plot (Zhou et al. 2007). The neutrality plot highlighted the coherence of mutation pressure and natural selection in framing CUB. In Figure 7, the graph was created by connecting the X-axis with GC3 and the Y-axis with GC12 (the sum of GC1 and GC2). If the RC value is ≥ 0.5, it indicates that mutational pressure has a stronger impact on the CUB of genes. We found that the slope of the regression line was 0.187, implying that mutational pressure contributed 18.7%, while natural selection contributed 81.3% in the CUB of Rhabdoviridae. Based on this observation, we concluded that the CUB of Rhabdoviridae was shaped mostly by natural selection. Our result was found comparable to the study of the Zika virus, which revealed that translational selection had played a major role in shaping the CUB of the Zika virus [51,56].
 | Figure 7: Neutrality plot of Rhabdoviridae (Regression coefficient value is 0.187 indicating that natural selection had a larger and more obvious impact, whereas mutational pressure had a considerably less impact in the codon usage pattern). [Click here to view] |
3.10. dN/dS Ratio and Evolutionary Research
The dN/dS ratio determines the mode and strength of selection by comparing synonymous substitution rates (dS) with non-synonymous substitution rates (dN). We performed a gene-wise dN/dS analysis for the Rhabdoviridae family to determine the magnitude of selection pressure affecting the gene encoding a protein. If dN/dS > 1, it indicates positive selection, which means that the encoded protein’s amino acid changes are supported by nature. Again, dN/dS < 1 reveals negative purifying selection where nature prevents amino acid changes, whereas dN/dS = 1 points toward neutral selection [57]. We found that the dN/dS ratio of the glycoprotein gene was 0.099, the matrix protein gene was 0.085, the nucleoprotein gene was 0.067, the phosphoprotein gene was 0.131, and the polymerase gene was 0.060. In our study, we found that the gene-wise dN/dS values were <1. Hence, the genes of the Rhabdoviridae family had undergone negative purifying selection to maintain their protein functioning in those viruses. A similar result (dN/dS < 1) was found in the study of BRCA1 gene of Homosapiens, Pongo pygmaeus, Nomascus leucogenys, and Nomascus gabriellae, suggesting that BRCA1 gene has experienced negative purifying selection [58].
Virus genomic sequence data can significantly contribute to the study of viral evolution and diversity among species. The phylogenetic studies emphasize the evolutionary distances and relationships between genes and genomes. To better comprehend the evolutionary relationships among Rhabdoviridae members, a phylogenetic tree based on the maximum likelihood method with a bootstrap value of 1000 was constructed using the Tamura Nei model [Figure 8]. In our study, the tree showed that closely related viruses were clearly divided into various clades. The majority of clades were significantly supported by high bootstrap scores (BS) and posterior probability (PP). There was evidence of a close relationship between the Kolente virus and Fikirini bat rhabdovirus (BS = 100; PP = 100), Lagos bat virus and Shimoni bat virus (BS = 100; PP = 100), and European bat lyssavirus 1 and Irkut virus (BS = 98; PP = 100). This evolutionary lineage of Rhabdoviridae family members might have resulted from both mutational pressure and natural selection. A recent study reported that distinct viral genotypes in the Lyssavirus genus, as well as in numerous species of vesiculoviruses and ephemeroviruses, were characterized using percentage sequence similarity within the nucleoprotein gene [59,60]. The evolutionary distance was computed using the Maximum likelihood method. From this analysis, we found that some members of Rhabdoviridae were closely associated and some were distantly related to each other. From the phylogenetic analysis of rabies virus, it was suggested that various species of bats in the United States were affected by the majority of diverse clades of rabies virus [61]. The extensive phylogenetic linkages across the Rhabdoviridae family and genera suggested that the evolutionary history of rhabdoviruses was substantially impacted by the method of transmission, host species (mammal, fish, or plant), and vector (homopteran, dipteran, or orthopteran) [62]. In this study, we found that the Lyssavirus genus separated apart from the other rhabdoviruses. Our findings were similar to those of the study on phylogenetic relationships among rhabdoviruses [62]. They suggested that the most obvious theory for these variations might be that the other genera are much older than the lyssaviruses. Furthermore, it was proposed that the extreme selection pressure against sequence change exhibited by lyssaviruses might potentially contribute to the limitation of the number of amino acids [62-64].
 | Figure 8: Gene-wise phylogenetic tree of Rhabdoviridae: (a) Glycoprotein gene, (b) Matrix protein gene, (c) Phosphoprotein gene, (d) Nucleoprotein gene, (e) Polymerase gene, and (f) L protein gene. [Click here to view] |
4. CONCLUSION
According to our findings, the highest frequency of nucleotide A was found in Rhabdoviridae CDSs. The majority of the codons ended with the T nucleotide. From the analysis of ENC values, it was observed that the genes of bat viruses under the Rhabdoviridae family had low CUB. In this study, we found that natural selection and mutation pressure, as progressive evolutionary factors, were both prominently featured in shaping the CUB of Rhabdoviridae. In addition, natural selection had a larger and more obvious impact, whereas mutational pressure had considerably less impact on the codon usage pattern of the 15 viral transcriptomes. Our findings revealed that the nucleotide base compositions were used to study the CUB of genes in Rhabdoviridae. CUB study might be useful in studying the virus-host relationship. It also summarizes molecular information, expressing gene factors, as well as the evolutionary processes of genes or genomes. The CUB study can be used as a starting point for developing vaccines against lethal viruses. This present study clarified and improved our understanding of the fundamental range of factors that influence the CUB of Rhabdoviridae. As a result, our research might usher in developing broad-spectrum resistance and treatments against the viruses of the Rhabdoviridae family.
5. ACKNOWLEDGMENTS
We are thankful to Assam University, Silchar, Assam, India, for providing the necessary facilities to carry out this work.
6. AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
7. FUNDING
There is no funding to report.
8. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
9. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
10. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
11. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
12. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Olival KJ, Hayman DT. Filoviruses in bats:Current knowledge and future directions. Viruses 2014;6:1759-88. [CrossRef]
2. Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg Infect Dis 2013;19:1819-23. [CrossRef]
3. Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, et al. Fruit bats as reservoirs of Ebola virus. Nature 2005;438:575-6. [CrossRef]
4. Amman BR, Albariño CG, Bird BH, Nyakarahuka L, Sealy TK, Balinandi S, et al. A recently discovered pathogenic Paramyxovirus, Sosuga virus, is present in Rousettus aegyptiacus fruit bats at multiple locations in Uganda. J Wildl Dis 2015;51:774-9. [CrossRef]
5. Mollentze N, Streicker DG. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc Natl Acad Sci U S A 2020;117:9423-30. [CrossRef]
6. Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017;546:646-50. [CrossRef]
7. Kuzmin IV, Novella IS, Dietzgen RG, Padhi A, Rupprecht CE. The rhabdoviruses:Biodiversity, phylogenetics, and evolution. Infect Genet Evol 2009;9:541-53. [CrossRef]
8. Walker PJ, Dietzgen RG, Joubert DA, Blasdell KR. Rhabdovirus accessory genes. Virus Res 2011;162:110-25. [CrossRef]
9. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010:A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2095-128. [CrossRef]
10. Wellehan JF Jr., Pessier AP, Archer LL, Childress AL, Jacobson ER, Tesh RB. Initial sequence characterization of the rhabdoviruses of squamate reptiles, including a novel rhabdovirus from a caiman lizard (Dracaena guianensis). Vet Microbiol 2012;158:274-9. [CrossRef]
11. Wang Y, Walker PJ. Adelaide river rhabdovirus expresses consecutive glycoprotein genes as polycistronic mRNAs:New evidence of gene duplication as an evolutionary process. Virology 1993;195:719-31. [CrossRef]
12. Allison AB, Mead DG, Palacios GF, Tesh RB, Holmes EC. Gene duplication and phylogeography of North American members of the Hart Park serogroup of avian rhabdoviruses. Virology 2014;448:284-92. [CrossRef]
13. Simon-Loriere E, Holmes EC. Gene duplication is infrequent in the recent evolutionary history of RNA viruses. Mol Biol Evol 2013;30:1263-9. [CrossRef]
14. Walker PJ, Byrne KA, Riding GA, Cowley JA, Wang Y, McWilliam S. The genome of bovine ephemeral fever rhabdovirus contains two related glycoprotein genes. Virology 1992;191:49-61. [CrossRef]
15. Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res 2003;92:1-7. [CrossRef]
16. Wong EH, Smith DK, Rabadan R, Peiris M, Poon LL. Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol Biol 2010;10:253. [CrossRef]
17. Ma QP, Li C, Wang J, Wang Y, Ding ZT. Analysis of synonymous codon usage in FAD7 genes from different plant species. Genet Mol Res 2015;14:1414-22. [CrossRef]
18. Liu Y. A code within the genetic code:Codon usage regulates co-translational protein folding. Cell Commun Signal 2020;18:145. [CrossRef]
19. Hooper SD, Berg OG. Gradients in nucleotide and codon usage along Escherichia coli genes. Nucleic Acids Res 2000;28:3517-23. [CrossRef]
20. Plotkin JB, Kudla G. Synonymous but not the same:The causes and consequences of codon bias. Nat Rev Genet 2011;12:32-42. [CrossRef]
21. Ingvarsson PK. Molecular evolution of synonymous codon usage in Populus. BMC Evol Biol 2008;8:307. [CrossRef]
22. Liu Q. Mutational bias and translational selection shaping the codon usage pattern of tissue-specific genes in rice. PLoS One 2012;7:e48295. [CrossRef]
23. Mazumdar P, Binti Othman R, Mebus K, Ramakrishnan N, Ann Harikrishna J. Codon usage and codon pair patterns in non-grass monocot genomes. Ann Bot 2017;120:893-909. [CrossRef]
24. Quax TE, Claassens NJ, Söll D, Van der Oost J. Codon bias as a means to fine-tune gene expression. Mol Cell 2015;59:149-61. [CrossRef]
25. Athey J, Alexaki A, Osipova E, Rostovtsev A, Santana-Quintero LV, Katneni U, et al. A new and updated resource for codon usage tables. BMC Bioinformatics 2017;18:391. [CrossRef]
26. Zhou Z, Dang Y, Zhou M, Li L, Yu CH, Fu J, et al. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc Natl Acad Sci U S A 2016;113:E6117-25. [CrossRef]
27. Gupta SK, Ghosh TC. Gene expressivity is the main factor in dictating the codon usage variation among the genes in Pseudomonas aeruginosa. Gene 2001;273:63-70. [CrossRef]
28. Zhou JH, Zhang J, Sun DJ, Ma Q, Chen HT, Ma LN, et al. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLoS One 2013;8:e77239. [CrossRef]
29. Wright F. The 'effective number of codons'used in a gene. Gene 1990;87:23-9. [CrossRef]
30. Sueoka N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J Mol Evol 1995;40:318-25. [CrossRef]
31. Shields DC, Sharp PM. Synonymous codon usage in Bacillus subtilis reflects both translational selection and mutational biases. Nucleic Acids Res 1987;15:8023-40. [CrossRef]
32. Sueoka N. Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci U S A 1988;85:2653-7. [CrossRef]
33. Gouy M, Gautier C. Codon usage in bacteria:Correlation with gene expressivity. Nucleic Acids Res 1982;10:7055-74. [CrossRef]
34. Gatherer D, McEwan NR. Small regions of preferential codon usage and their effect on overall codon bias--the case of the plp gene. Biochem Mol Biol Int 1997;43:107-14. [CrossRef]
35. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X:Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018;35:1547-9. [CrossRef]
36. Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 1993;10:512-26.
37. Hammer Ø, Harper DA, Ryan PD. PAST:Paleontological statistics software package for education and data analysis. Palaeontol Electron 2001;4:9.
38. Kosakovsky Pond SL, Frost SD. Not so different after all:A comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 2005;22:1208-22. [CrossRef]
39. Kumar S, Stecher G, Tamura K. MEGA7:Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 2016;33:1870-4. [CrossRef]
40. Nie X, Deng P, Feng K, Liu P, Du X, You FM, et al. Comparative analysis of codon usage patterns in chloroplast genomes of the Asteraceae family. Plant Mol Biol Rep 2014;32:828-40. [CrossRef]
41. Van Hemert F, Van der Kuyl AC, Berkhout B. Impact of the biased nucleotide composition of viral RNA genomes on RNA structure and codon usage. J Gen Virol 2016;97:2608-19. [CrossRef]
42. Deb B, Uddin A, Chakraborty S. Codon usage pattern and its influencing factors in different genomes of hepadnaviruses. Arch Virol 2020;165:557-70. [CrossRef]
43. Rajewska M, Wegrzyn K, Konieczny I. AT-rich region and repeated sequences-the essential elements of replication origins of bacterial replicons. FEMS Microbiol Rev 2012;36:408-34. [CrossRef]
44. Wan XF, Xu D, Kleinhofs A, Zhou J. Quantitative relationship between synonymous codon usage bias and GC composition across unicellular genomes. BMC Evol Biol 2004;4:19. [CrossRef]
45. Huang X, Xu J, Chen L, Wang Y, Gu X, Peng X, et al. Analysis of transcriptome data reveals multifactor constraint on codon usage in Taenia multiceps. BMC Genomics 2017;18:308. [CrossRef]
46. Dutta R, Buragohain L, Borah P. Analysis of codon usage of severe acute respiratory syndrome corona virus 2 (SARS-CoV-2) and its adaptability in dog. Virus Res 2020;288:198113. [CrossRef]
47. Wang L, Xing H, Yuan Y, Wang X, Saeed M, Tao J, et al. Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS One 2018;13:e0194372. [CrossRef]
48. Zhou T, Gu W, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses. Biosystems 2005;81:77-86. [CrossRef]
49. Chakraborty S, Deb B, Barbhuiya PA, Uddin A. Analysis of codon usage patterns and influencing factors in Nipah virus. Virus Res 2019;263:129-38. [CrossRef]
50. Kumar N, Bera BC, Greenbaum BD, Bhatia S, Sood R, Selvaraj P, et al. Revelation of influencing factors in overall codon usage bias of equine influenza viruses. PLoS One 2016;11:e0154376. [CrossRef]
51. Wang H, Liu S, Zhang B, Wei W. Analysis of synonymous codon usage bias of Zika virus and its adaption to the hosts. PLoS One 2016;11:e0166260. [CrossRef]
52. Gu W, Zhou T, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the nidovirales. Virus Res 2004;101:155-61. [CrossRef]
53. Sueoka N. Correlation between base composition of deoxyribonucleic acid and amino acid composition of protein. Proc Natl Acad Sci U S A 1961;47:1141-9. [CrossRef]
54. Cheng X, Virk N, Chen W, Ji S, Ji S, Sun Y, et al. CpG usage in RNA viruses:Data and hypotheses. PLoS One 2013;8:e74109. [CrossRef]
55. Vicario S, Moriyama EN, Powell JR. Codon usage in twelve species of Drosophila. BMC Evol Biol 2007;7:226. [CrossRef]
56. Liu Q. Analysis of codon usage pattern in the radioresistant bacterium Deinococcus radiodurans. Biosystems 2006;85:99-106. [CrossRef]
57. Anisimova M, Liberles DA. The quest for natural selection in the age of comparative genomics. Heredity (Edinb) 2007;99:567-79. [CrossRef]
58. Mazumder TH, Uddin A, Chakraborty S. Transcription factor gene GATA2:Association of leukemia and nonsynonymous to the synonymous substitution rate across five mammals. Genomics 2016;107:155-61. [CrossRef]
59. Arai YT, Kuzmin IV, Kameoka Y, Botvinkin AD. New Lyssavirus genotype from the lesser mouse-eared bat (Myotis blythii), Kyrghyzstan. Emerg Infect Dis 2003;9:333-7. [CrossRef]
60. Kuzmin IV, Orciari LA, Arai YT, Smith JS, Hanlon CA, Kameoka Y, et al. Bat Lyssaviruses (Aravan and Khujand) from Central Asia:Phylogenetic relationships according to N, P and G gene sequences. Virus Res 2003;97:65-79. [CrossRef]
61. Hyeon JY, Risatti GR, Helal ZH, McGinnis H, Sims M, Hunt A, et al. Whole genome sequencing and phylogenetic analysis of rabies viruses from bats in Connecticut, USA, 2018-2019. Viruses 2021;13:2500. [CrossRef]
62. Bourhy H, Cowley JA, Larrous F, Holmes EC, Walker PJ. Phylogenetic relationships among rhabdoviruses inferred using the L polymerase gene. J Gen Virol 2005;86:2849-58. [CrossRef]
63. Guyatt KJ, Twin J, Davis P, Holmes EC, Smith GA, Smith IL, et al. A molecular epidemiological study of Australian bat Lyssavirus. J Gen Virol 2003;84:485-96. [CrossRef]
64. Holmes EC, Woelk CH, Kassis R, Bourhy H. Genetic constraints and the adaptive evolution of rabies virus in nature. Virology 2002;292:247-57. [CrossRef]